Validating Self-Produced Sensors: A Size-Exclusion Chromatography Guide for Biopharmaceutical Analysis
This article provides a comprehensive framework for researchers and drug development professionals seeking to validate self-produced sensors using Size-Exclusion Chromatography (SEC).
Validating Self-Produced Sensors: A Size-Exclusion Chromatography Guide for Biopharmaceutical Analysis
Abstract
This article provides a comprehensive framework for researchers and drug development professionals seeking to validate self-produced sensors using Size-Exclusion Chromatography (SEC). It covers foundational SEC principles for protein analysis and aggregate detection, outlines methodological approaches for integrating novel sensors into SEC workflows, addresses common troubleshooting scenarios, and establishes rigorous validation protocols against orthogonal methods. By bridging innovative sensor technology with established chromatographic techniques, this guide aims to support the development of reliable, in-house analytical tools for characterizing biopharmaceuticals, from monoclonal antibodies to gene therapy vectors like AAVs, ultimately enhancing process development and quality control.
Core Principles of Size-Exclusion Chromatography in Biomolecular Analysis
Size-exclusion chromatography (SEC) has established itself as a critical analytical technique for the separation and characterization of macromolecules based on their size in solution. For researchers validating self-produced sensors, understanding the performance characteristics of the SEC stationary phase is fundamental, as it directly impacts the reproducibility, accuracy, and reliability of the separation data. The journey of SEC stationary phases from simple soft gels to advanced hybrid particles represents a continuous pursuit of improved efficiency, stability, and minimal analyte interaction. This evolution has been particularly crucial in the biopharmaceutical industry, where the quantitative assessment of protein aggregates is essential due to their potential effects on product efficacy and immunogenicity [1]. This guide provides a comprehensive comparison of SEC stationary phases, equipping scientists with the knowledge to select the appropriate phase for their specific application, including method validation for novel sensor technologies.
The Historical Progression of SEC Stationary Phases
The development of SEC stationary phases has traversed several distinct generations, each marked by significant improvements in material science and a deeper understanding of separation mechanics. The timeline below illustrates the key milestones in this evolutionary pathway.
The conceptual foundation for size-based separations was first established in the 1950s with the use of starch and early gels as packing materials. Lindqvist and Storgårds reported the first separation of biomolecules using starch, and Lathe and Ruthven further demonstrated the separation of proteins and peptides using maize starch, describing it as a "molecular sieve" effect [1]. While revolutionary, these materials suffered from low mechanical strength, leading to bed collapse at high linear velocities [1]. The 1960s saw the introduction of cross-linked dextrans, commercialized as Sephadex, which offered greater mechanical strength and became the standard for size-based protein separations for many years [1]. By varying the degree of cross-linking, manufacturers could control the pore size and thus the separation range. Other polymeric resins like polyacrylamide (Bio-Gel) and agarose were also introduced during this period [1] [2]. Despite these improvements, the soft polymeric resins remained prone to compression under pressure, limiting further reductions in particle size [1].
A significant leap occurred in the 1970s with the adoption of derivatized porous silica. Its superior mechanical strength and non-swelling nature allowed for the use of smaller particles, thereby enhancing chromatographic efficiency [1]. However, a major drawback was the strong ionic interaction between acidic surface silanols and proteins, which necessitated surface modifications and mobile phase additives [1]. Early modifiers included glyceropropylsilane and N-acetylaminopropylsilane, but these often introduced undesirable hydrophobic interactions with proteins [1]. The most successful surface modifier for silica, the diol functional group, was introduced during this era and is still valued for its minimal hydrophobic interactions [1]. The first-generation hybrid particles, introduced in 1999, represented a paradigm shift by combining organic and inorganic components [3]. Synthesized from tetraethoxysilane and methyltriethoxysilane, these methyl hybrid particles exhibited improved stability in alkaline mobile phases and reduced silanol activity compared to pure silica, resulting in better peak shapes for basic analytes [3]. Their main limitation was insufficient mechanical strength for the ultra-high pressures used in modern UHPLC systems [3]. This was addressed in 2004 with the second-generation hybrid particles, known as Bridged-Ethylene Hybrid (BEH) technology [1] [3]. These particles, synthesized using bridged silane precursors like 1,2-bis(triethoxysilyl)ethane, featured a more robust structure without sacrificing the beneficial hybrid properties. This made them suitable for smaller particle sizes (down to 1.7 μm) and capable of withstanding the high pressures (>6000 psi) of UHPLC systems, enabling faster and more efficient separations [1] [3].
Comparative Analysis of Stationary Phase Materials
Table 1: Performance Characteristics of Different SEC Stationary Phase Materials
| Material Type | Relative Mechanical Strength | pH Stability Range | Key Advantages | Primary Limitations | Typical Applications |
|---|---|---|---|---|---|
| Soft Gels (Dextran, Agarose) | Low | 2-10 [1] | Minimal protein adsorption; high porosity for large biomolecules | Low pressure tolerance; bed compression; slow flow rates | Desalting; protein purification; separation of large biomolecules |
| Rigid Silica | High | 2-8 [3] | High efficiency; small particles possible; stable at high pressures | Strong ionic interactions with silanols; limited pH stability | Analysis of synthetic polymers (in organic solvents) |
| Diol-Modified Silica | High | 2-8 | Reduced hydrophobic interactions; improved peak shape vs. bare silica | Requires high ionic strength mobile phases to mask residual silanols | Standard SEC analysis of proteins and antibodies |
| 1st Gen Methyl Hybrid | Medium | 2-12 [3] | Reduced silanol activity; improved peak shape for bases; wider pH range than silica | Limited mechanical stability for UHPLC pressures | Method development for ionizable analytes; preparative purifications |
| 2nd Gen BEH Hybrid | High | 1-12 [3] | Superior mechanical strength; extended pH stability; high efficiency at UHPLC pressures | Higher cost than traditional silica | High-resolution SEC for proteins and mAbs; oligonucleotide analysis; UHPLC applications |
Table 2: Key Experimental Parameters for SEC Stationary Phase Characterization
| Characterization Parameter | Experimental Protocol | Soft Gels | Diol-Milica | BEH Hybrid |
|---|---|---|---|---|
| Column Efficiency (Plate Count, N/m) | Inject a small, unretained molecule (e.g., acetone). Calculate N = 5.54 *(tR/w1/2)2, where tR is retention time and w1/2 is peak width at half height. Normalize for column length [4]. | Low | Medium-High | High |
| Resolution (Rs) | Inject a protein mixture (e.g., monomer/dimer). Calculate Rs = 2*(tR2-tR1)/(w1+w2), where w is the peak width at baseline [1]. | Moderate | Good | Excellent |
| Silanol Activity (Peak Asymmetry for Bases) | Inject a basic analyte (e.g., lidocaine) at neutral pH. Measure the peak asymmetry factor (As). A value of 1.0 indicates a symmetric peak [3]. | Not Applicable | Moderate (As > 1.2) | Low (As ≈ 1.0) |
| Pressure Tolerance | Gradually increase flow rate and record backpressure. Note the point of significant bed compression or pressure instability. | Low (< 500 psi) | High (> 6000 psi) | Very High (≥ 15,000 psi) |
| Aggregate Recovery | Inject a stressed protein sample and compare the peak areas of aggregates and monomers to a reference standard. Higher recovery indicates less surface adsorption [1] [5]. | High | Moderate-High | High |
The data in Table 1 and Table 2 highlight the clear performance trajectory from soft gels to modern hybrids. The primary challenge with early soft gels was their mechanical weakness, restricting their use to low-pressure systems and resulting in longer analysis times [1] [6]. The introduction of rigid silica was a breakthrough for efficiency and speed but introduced significant surface reactivity. Acidic silanols on the silica surface could cause ionic interactions with basic analytes, leading to peak tailing and inaccurate quantification—a critical issue when characterizing protein aggregates or using SEC for sensor validation [1] [3]. Diol-modified silica phases mitigated the hydrophobic interaction issue but still required mobile phase additives (e.g., salts at concentrations of 150 mM or higher) to suppress residual ionic interactions [1].
The advent of hybrid particle technology addressed these core limitations. The incorporation of organic moieties directly into the particle skeleton significantly reduced the number and acidity of surface silanols. This is evidenced by the superior peak symmetry (As ≈ 1.0) for basic analytes run at neutral pH, as shown in Table 2 [3]. Furthermore, the hybrid structure confers exceptional stability across a broad pH range (pH 1-12), providing chromatographers with a powerful tool for method development. The ability to operate at high pH can increase loading capacity for preparative applications and offers a unique selectivity parameter for separating ionizable analytes [3]. The second-generation BEH hybrid particles combined this superior chemistry with the mechanical strength required for UHPLC, enabling the use of sub-2-μm particles for fast, high-resolution separations without compromising column longevity [1] [3].
Essential Research Reagent Solutions for SEC Analysis
Table 3: Key Reagents and Materials for SEC Experiments
| Item | Function/Description | Application Example |
|---|---|---|
| Diol-Modified Silica Columns | Standard SEC columns with hydrophilic diol bonding; provide a good balance of performance and cost. | Routine analysis of protein aggregates and mAb monomers [1]. |
| BEH Hybrid SEC Columns | Advanced columns with organic-inorganic hybrid structure; offer high efficiency, low interaction, and wide pH stability. | High-resolution, high-speed SEC in UHPLC systems; analysis of sensitive biomolecules [1] [3]. |
| Mobile Phase Buffers | Aqueous buffers (e.g., phosphate, Tris) with added salt (150-250 mM) to mask residual surface charges on the stationary phase. | Essential for preventing ionic interactions and achieving true size-based separation in aqueous SEC [1] [5]. |
| Molecular Weight Standards | Monodisperse proteins or polymers with known molecular weights; used for column calibration. | Creating a calibration curve of log(MW) vs. retention volume to estimate sample molecular weight [1] [7]. |
| Multi-Angle Light Scattering (MALS) Detector | An absolute detector that measures molecular weight independently of elution volume, eliminating the need for column calibration. | Determining absolute molecular weight and detecting aggregates in IgG-HRP conjugation reactions [5] [8]. |
Advanced Detection and Validation Strategies in SEC
For researchers validating self-produced sensors, employing advanced detection methods is crucial for obtaining absolute data that is not reliant on column calibration. The integration of multiple detectors provides a comprehensive characterization of the sample and the separation process. The workflow below outlines how these detectors function together in a multi-detection SEC system.
As illustrated, a refractive index (RI) detector is a concentration-sensitive bulk property detector, while a UV detector is a solute property detector that is particularly useful for proteins [2] [5]. The key advancement for absolute validation is the inclusion of a light scattering (LS) detector (MALS or RALS/LALS), which measures molecular weight directly at each elution slice without relying on calibration standards or retention time [5] [8]. This is critical for validating the performance of SEC columns and sensors, as it removes shape-dependent inaccuracies that can occur with conventional calibration. A viscometer detector provides intrinsic viscosity data, offering insights into macromolecular structure, conformation, and branching [5] [8].
The power of this multi-detection approach is exemplified in its application for monitoring a conjugation reaction, such as the preparation of an IgG-HRP conjugate. A 2023 study used multi-detection SEC to characterize starting materials, intermediates, and the final product in detail [5]. The methodology provided absolute molecular weight values (~153 kDa for IgG monomer, ~43 kDa for HRP, and ~235 kDa for the successful conjugate), polydispersity (Mw/Mn ≈ 1.003 for IgG, indicating homogeneity), and aggregate content for each species [5]. This level of characterization ensures that the conjugation process is accurately monitored and the final product is properly validated—a principle that applies directly to the validation of SEC-based sensors.
The evolution of SEC stationary phases, from the soft, compressible beds of starch to the robust, high-performance diol-modified hybrid particles, has been driven by the need for greater efficiency, speed, and most importantly, separation fidelity. For scientists engaged in the validation of self-produced sensors, the choice of stationary phase is not merely a technical detail but a fundamental decision that influences the accuracy of all subsequent data. Modern BEH hybrid particles with diol surfaces currently represent the state-of-the-art, offering minimal interaction, broad pH stability, and the mechanical strength required for high-resolution separations. When coupled with advanced detection methods like multi-angle light scattering, researchers possess a powerful toolkit for obtaining absolute molecular parameters, ensuring that their separations are based solely on size and that their sensors are validated against the most rigorous standards.
Size-exclusion chromatography (SEC), also historically known as gel-filtration chromatography or molecular sieve chromatography, is a fundamental technique for separating biomolecules based on their size in solution [1]. Unlike other chromatographic methods that rely on chemical interactions, SEC operates on a unique entropy-driven principle, making it particularly valuable for analyzing the native states of proteins, protein aggregates, and other large biomolecules without altering their structure [1]. For researchers validating self-produced sensors, SEC serves as a critical orthogonal method, providing a benchmark for size-based characterization due to its well-understood mechanism and reproducible results.
The Entropy-Driven Mechanism of SEC
Theoretical Foundation
The separation mechanism in SEC is fundamentally different from other chromatographic modes. In most chromatography, the enthalpy of adsorption (ΔH) is the dominant contributor to the overall change in free energy. SEC is unique because partitioning is driven almost entirely by entropic processes with no significant adsorption enthalpy [1].
The thermodynamic principle can be described by the equation:
ΔG⁰ = -RTlnK = ΔH⁰ - TΔS⁰
Where ΔG⁰ is the standard free energy change, R is the gas constant, T is absolute temperature, and K is the partition coefficient. For ideal SEC separations, ΔH = 0, simplifying the equation to:
ΔG⁰ = -TΔS⁰
This reveals that the driving force for separation is purely entropic (-TΔS⁰) [1]. The thermodynamic retention factor (K_D) represents the fraction of intraparticle pore volume accessible to the analyte:
KD = (VR - V0)/Vi
Where VR is the retention volume of the analyte, V0 is the interstitial volume, and Vi is the intra-particle volume [1]. KD ranges from 0 (analyte fully excluded from pores) to 1 (analyte fully accesses all pores).
Separation Process Visualization
Historical Development and Materials
The concept of size-based separations was first speculated by Synge and Tiselius, based on observations that small molecules could be excluded from zeolite pores [1]. The term "molecular sieve" was coined by J.W. McBain to describe this property [1]. The first separation of biomolecules by SEC was reported by Lindqvist and Storgårds, who separated peptides from amino acids on a column packed with starch [1]. Lathe and Ruthven subsequently performed extensive characterizations using potato or maize starch, demonstrating the separation of various compounds including proteins and peptides by the molecular sieve effect [1].
Modern SEC materials have evolved significantly:
- Dextran-based gels (Sephadex): Crosslinked with epichlorohydrin, providing greater mechanical strength than starch [1]
- Polyacrylamide gels (Bio-Gel): Commercialized by Bio-Rad for size-based separations [1]
- Porous silica: Provides superior mechanical strength but requires surface modifications to minimize ionic interactions [1]
- Hybrid particles: BEH particles with diol groups reduce silanol activity and allow smaller particle sizes down to 1.7μm [1]
Comparative Analysis of SEC and Alternative Techniques
Performance Comparison Table
Table 1: Comparative analysis of SEC and alternative size-based separation techniques
| Technique | Separation Mechanism | Effective Size Range | Analysis Time | Key Applications | Resolution Limitations |
|---|---|---|---|---|---|
| Size-Exclusion Chromatography | Entropy-driven pore access | 1-1000 kDa [1] | 10-30 minutes [1] | Protein aggregation analysis, polymer separation [1] | Limited by column efficiency and pore volume [1] |
| Pulsed-Field Gel Electrophoresis | Biased reptation in alternating electric fields | >10 kbp DNA [9] | 1-2 days [9] | Bacterial genotyping, large DNA separation [9] | Time-consuming, risk of DNA fragmentation [9] |
| Nanoslit Relaxation Analysis | DNA relaxation dynamics in confinement | >10 kbp DNA [9] | ~60 seconds [9] | Rapid DNA sizing, genotyping [9] | Requires specialized nanofluidic fabrication [9] |
| Analytical Ultracentrifugation | Sedimentation under centrifugal force | Broad range | Several hours | Orthogonal validation for SEC [1] | Equipment intensive, low throughput [1] |
| Asymmetric Flow Field Flow Fractionation | Cross-flow separation in channel | 1 kDa - 100 μm | 30-60 minutes | Protein complexes, nanoparticles [1] | Method development complexity [1] |
Experimental Resolution Data
Table 2: Experimental resolution comparison for DNA separation methods
| Method | Resolution Metric | Analysis Time | Sample Requirements | Fragmentation Risk |
|---|---|---|---|---|
| Pulsed-Field Gel Electrophoresis | Standard reference method [9] | 24-48 hours [9] | Standard DNA preparation | High (repeated hooking and stretching) [9] |
| Nanoslit Relaxation (130 nm depth) | Partial separation of λ and T4 DNA [9] | ~60 seconds [9] | Minimal sample processing | Low (controlled stretching <30%) [9] |
| Nanoslit Relaxation (49 nm depth) | Resolution = 2.33 [9] | ~60 seconds [9] | Minimal sample processing | Very low [9] |
| Artificial Gel Nanostructures | Varies with constriction size [9] | 30 minutes [9] | Standard DNA preparation | Medium (frequent gel interactions) [9] |
Experimental Protocols and Methodologies
Standard SEC Protocol for Protein Analysis
Column Preparation and Selection:
- Use diol-modified silica or hybrid particles (e.g., BEH technology) for minimal protein interaction [1]
- Column dimensions typically 7.8mm ID × 300mm with 1.7-5μm particle sizes [1]
- Equilibrate with 5-10 column volumes of mobile phase before analysis
Mobile Phase Requirements:
- Use aqueous buffers (e.g., phosphate, Tris) with 100-200mM NaCl to minimize ionic interactions [1]
- Maintain pH between 6.0-7.5 for most protein applications
- Filter through 0.22μm membrane and degas before use
Sample Preparation:
- Centrifuge samples at 10,000-15,000g for 10 minutes to remove particulates
- Match sample buffer composition to mobile phase to avoid viscosity artifacts
- Ideal injection volume: 1-5% of column volume for optimal resolution
Chromatographic Conditions:
- Flow rate: 0.5-1.0mL/min for analytical columns (4.6-7.8mm ID)
- Temperature: 15-25°C (minimal temperature effect on retention) [1]
- Detection: UV 280nm for proteins, or TOC detection for comprehensive analysis [10]
SEC Validation Methodology
Linearity and Range:
- Establish linear range using protein standards of known molecular weight
- Typical linear range: 1-10,000 μgC dm⁻³ for TOC detection [10]
- Correlation coefficient (R²) should exceed 0.995 for quantitative work
Precision and Accuracy:
- Repeatability: %RSD <2% for retention time, <5% for peak area [10]
- Intermediate precision: %RSD <5% over multiple days with different columns
- Accuracy: 85-115% recovery for known standards [10]
Limit of Detection and Quantification:
- LOD: Typically 19.0 μgC dm⁻³ for TOC detection [10]
- LOQ: Typically 3-5 times LOD, depending on detection method
HPSEC-TOC Protocol for Natural Organic Matter
Sample Preservation:
- Refrigerate samples immediately after collection
- Maximum storage duration: Two weeks [10]
- Avoid freezing and thawing cycles which alter NOM composition
Inorganic Carbon Removal:
- Acidify samples to pH 6.0 with minimal hydrochloric acid
- Purge with N₂ gas for 5-10 minutes [10]
- Verify complete IC removal by measuring TOC before and after treatment
Chromatographic Conditions:
- Column: Preparative TSK HW-50S [10]
- Injection volume: 1350 mm³ [10]
- Mobile phase: Appropriate aqueous buffer matching sample ionic strength
- Detection: TOC via UV/persulfate oxidation [10]
Research Reagent Solutions and Essential Materials
Table 3: Essential research reagents and materials for SEC analysis
| Material/Reagent | Function/Purpose | Key Characteristics | Example Applications |
|---|---|---|---|
| Diol-Modified Silica Columns | Size-based separation matrix | High mechanical strength, minimal protein interaction [1] | Protein aggregation analysis, monoclonal antibody characterization [1] |
| Hybrid BEH Particles | Advanced SEC stationary phase | Reduced silanol activity, 1.7μm particle size [1] | High-resolution protein separation [1] |
| TSK HW-50S Column | Semipreparative SEC separation | Optimized for natural organic matter [10] | HPSEC-TOC analysis of freshwater NOM [10] |
| Aqueous SEC Buffers | Mobile phase for biomolecules | 100-200mM NaCl, pH 6.0-7.5 [1] | Maintaining protein stability during analysis [1] |
| Protein Molecular Weight Standards | Column calibration and validation | Broad molecular weight range (1-670 kDa) [1] | Creating calibration curves, method validation [1] |
| TOC Detection System | Universal carbon detection | UV/persulfate oxidation method [10] | Comprehensive NOM analysis, non-UV absorbing compounds [10] |
Technological Advances and Future Directions
Recent advancements in SEC technology have focused on improving resolution, speed, and applicability to various biomolecules. The development of sub-2μm particles using hybrid technology has significantly enhanced chromatographic efficiency while maintaining mechanical stability [1]. The integration of multiple detection systems including TOC, UV, and light scattering provides comprehensive characterization in a single analysis [10].
For researchers validating self-produced sensors, SEC continues to provide the gold standard for size-based characterization due to its robust theoretical foundation, reproducibility, and minimal sample alteration during analysis. The entropy-driven mechanism ensures that biomolecules remain in their native state, providing confidence in size distribution measurements critical for sensor validation studies.
The experimental data demonstrates that while newer techniques like nanoslit relaxation analysis offer dramatic improvements in speed for specific applications like DNA analysis [9], SEC maintains its position as the most versatile and widely applicable method for broad-spectrum biomolecule separation and characterization.
In biopharmaceutical development, robust analytical methods are essential for characterizing protein therapeutics, ensuring their safety, efficacy, and quality. Protein aggregation can impact economic viability, reduce bioactivity, and increase immunogenicity because the recipient's immune system may recognize the protein complex as nonself [11]. Similarly, accurate determination of molecular weight (MW) and purity is critical, as these attributes influence biological activity and stability. This guide compares key analytical technologies, focusing on the role of Size-Exclusion Chromatography (SEC) as a reference method for validating novel sensor-based approaches, such as biosensors and process analytical technology (PAT) tools [12] [13].
Technology Performance Comparison
The following analytical techniques are central to monitoring critical quality attributes (CQAs) in biopharmaceuticals. Their performance characteristics are summarized in Table 1.
Table 1: Comparison of Analytical Techniques for Protein Characterization
| Technique | Key Applications | Key Performance Metrics | Throughput & Ease of Use | Primary Limitations |
|---|---|---|---|---|
| Size-Exclusion Chromatography (SEC) | Soluble aggregate analysis and quantitation [11]; Oligomeric state determination [14]. | Robust and accurate for soluble aggregates (1–50 nm) [11]; Resolution depends on column pore size and mobile phase [11] [14]. | Requires skilled personnel and laboratory infrastructure; Can be automated for quality control [11]. | Risk of shear forces or column interactions altering sample [11]; Mobile phase composition significantly influences elution [14]. |
| Light Scattering (SLS/DLS) | Direct measurement of absolute molecular weight [14]; Estimation of second virial coefficient (B22) for aggregation propensity [14]. | Provides volume-averaged molar masses without calibration curves [14]; Sensitive to presence of large aggregates [14]. | Requires specific expertise for data interpretation. | Results are sensitive to sample quality and solution conditions. |
| Process Analytical Technology (PAT) e.g., NIR, Raman Spectroscopy | Real-time monitoring of CPPs and CQAs [13]; In-line monitoring of tablet content uniformity [13]. | Enables proactive quality control and continuous process verification [13]. | High throughput; Suitable for in-line/on-line deployment in manufacturing [13]. | Requires complex model calibration and validation [13]; Data integration challenges [13]. |
| Biosensors | Detection of contaminants, pathogens, and additives [12]; Potential for intelligent packaging [12]. | High sensitivity and specificity [12]; Rapid response times [12]. | Amenable to portability and on-site use [12]. | Susceptible to matrix effects in complex samples [12]; Requires standardization for regulatory acceptance [12]. |
| Cyclic Voltammetry (CV) | Polymer molecular weight analysis via solution viscosity correlation [15]. | Provides results within seconds [15]. | Rapid measurement; Minimal sample preparation [15]. | Primarily demonstrated for synthetic polymers; applicability to proteins may be limited. |
Experimental Protocols for Key Applications
Protocol 1: Monitoring Protein Aggregation via SEC
SEC separates molecules based on their hydrodynamic size, making it ideal for resolving monomeric proteins from soluble aggregates like dimers and higher-order oligomers [11].
Detailed Methodology:
- Column Selection: Use an SEC column with a pore size appropriate for the target protein's hydrodynamic radius. Columns with 150-300 Å pores are common for many proteins. Connecting columns in series can increase resolution [11].
- Mobile Phase Preparation: The mobile phase must preserve protein native state and prevent nonspecific interactions. A typical phase is 50 mM sodium phosphate, 150 mM sodium chloride, pH 6.8. Note that ionic strength and pH can significantly impact elution volume and must be controlled [11] [14].
- Sample Preparation: Filter the protein sample using a 0.22 µm membrane to remove particulates. The injection concentration should be below the critical concentration to avoid viscosity effects and "macromolecular crowding" [7].
- System Setup and Calibration: Use an HPLC system with a UV detector (e.g., 280 nm). Calibrate the column using a protein standard mix with known molecular weights (e.g., thyroglobulin, IgG, ovalbumin) to create a log(MW) vs. elution volume curve [11] [14].
- Chromatographic Run: Inject the sample and run isocratically. Monitor the elution profile to identify the monomer peak and earlier-eluting aggregate peaks.
- Data Analysis: Quantify the percentage of monomer and aggregates based on the relative peak areas. For absolute molecular weight determination without calibration, couple SEC in-line with a static light scattering (SLS) detector [14].
Protocol 2: Validating SEC Method Accuracy
Accuracy validation is crucial when SEC is used for determining molecular weight distributions (MWD) and averages.
Detailed Methodology:
- Prepare a Polydisperse Reference Standard: Create a mixture of two well-characterized, monodisperse protein standards with certified molecular weights (M1 and M2). The mixture should mimic the Mw and Mn of the sample. Using equal weights of the two standards simplifies calculations and minimizes purity-related errors [7].
- Calculate True Values: The true number-average (Mn)t and weight-average (Mw)t molecular weights of the mixture are calculated using the equations:
- (Mn)t = 2 / (1/M1 + 1/M2)
- (Mw)t = (M1 + M2) / 2 [7]
- Analysis and Accuracy Calculation: Analyze the prepared standard using the SEC method. Determine the experimental molecular weight averages, (Mn)exp and (Mw)exp. Calculate the absolute error (AE) and relative error (%RE) to validate accuracy:
- (Mn)AE = (Mn)exp - (Mn)t
- %(Mn)RE = [(Mn)AE / (Mn)t] × 100% [7]
Protocol 3: Real-time Monitoring with PAT
PAT tools like NIR spectroscopy enable real-time monitoring during manufacturing.
Detailed Methodology:
- Sensor Integration: Integrate an NIR probe directly into the process stream, such as a bioreactor or a conduit in a continuous manufacturing line [13].
- Data Acquisition: Collect NIR spectra in real-time. The spectra result from molecular overtones and combination vibrations of C-H, O-H, and N-H bonds, providing chemical and physical information [13].
- Chemometric Modeling: Use multivariate calibration models (e.g., partial least squares, PLS) to correlate spectral data with reference values (e.g., protein concentration, aggregation index) obtained from primary methods like SEC [13].
- Process Control: The real-time data is fed into a process control system to enable dynamic adjustment of Critical Process Parameters (CPPs), ensuring consistent product quality [13].
Workflow and Relationship Visualizations
SEC Method Validation Workflow
The following diagram illustrates the key stages in developing and validating a reliable SEC method.
PAT Integration for Real-Time Control
This diagram shows how PAT tools are integrated into a bioprocess for real-time monitoring and control.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials for SEC and Aggregation Analysis
| Item | Function | Key Considerations |
|---|---|---|
| SEC Columns | Separates molecules by hydrodynamic size [11]. | Pore size (e.g., 150 Å, 300 Å) must match target protein size. Columns can be used in series for increased resolution [11]. |
| Protein Standards | Calibrates SEC system for molecular weight determination [7] [11]. | Use monodisperse standards (e.g., thyroglobulin, IgG) or prepared mixtures to validate accuracy [7]. |
| Mobile Phase Buffers | Dissolves and elutes samples under controlled conditions. | Composition (pH, ionic strength) is critical for accuracy and must be consistent, as it affects protein elution and stability [11] [14]. |
| Static Light Scattering (SLS) Detector | Provides direct, absolute measurement of molecular weight in-line with SEC [14]. | Bypasses need for calibration curves; allows detection of aggregates and calculation of virial coefficients [14]. |
| Total Organic Carbon (TOC) Analyzer | Quantifies organic carbon in samples; used in specialized SEC for natural organic matter [16]. | Membrane-based conductivity provides specificity, meeting USP requirements for accuracy and validation [17]. |
Size-exclusion chromatography (SEC) is a pivotal analytical technique within the biopharmaceutical industry, enabling the separation of biomolecules based on their hydrodynamic size [18]. This gentle, solution-based method is indispensable for characterizing critical quality attributes (CQAs) of therapeutic proteins, including monoclonal antibodies, biosimilars, and newer modalities like antibody-drug conjugates (ADCs) and gene therapy products [19] [20]. As a quality assurance tool, SEC provides essential data on aggregation, fragmentation, and oligomeric distribution throughout the biopharma workflow—from upstream process development to final product quality control and regulatory assessments for biosimilar approval [21] [22]. The technique's ability to maintain molecular integrity during analysis makes it particularly valuable for monitoring product stability and process consistency, forming a critical component of the quality-by-design (QbD) framework mandated by regulatory agencies worldwide [23] [24].
Fundamental Principles and Methodological Advancements in SEC
Core Separation Mechanism
SEC operates on the principle of differential partitioning between a mobile phase and porous stationary phase [18]. As a sample mixture travels through the column, smaller molecules diffuse into the pores of the packing material, thereby traversing a longer path, while larger molecules are excluded from these pores and elute first [18]. This process separates molecules in order of decreasing hydrodynamic size, with the total volume of the mobile phase in the column determining the separation efficiency [18]. The selection of appropriate pore sizes in the stationary phase is crucial for achieving optimal resolution for target analytes, whether analyzing monoclonal antibody monomers and aggregates or characterizing complex gene therapy products like recombinant adeno-associated viruses (rAAVs) and mRNA [19].
Advanced SEC Platforms and Orthogonal Approaches
Recent technological innovations have significantly enhanced SEC capabilities. High-performance SEC (HPSEC) utilizing smaller particle sizes and higher pressures now provides improved resolution and faster analysis times [18]. The integration of multi-angle light scattering (MALS) detection with SEC allows for direct determination of molecular weight and size, independent of elution volume [22] [18]. Similarly, native SEC-mass spectrometry (nSEC-MS) has emerged as a powerful platform for characterizing labile biopharmaceuticals, enabling simultaneous separation and identification under non-denaturing conditions [20]. This approach has been successfully validated for quantifying critical attributes like drug-to-antibody ratio (DAR) and drug load distribution (DLD) in cysteine-linked antibody-drug conjugates [20].
The concept of orthogonality—using techniques with different physicochemical principles to assess the same attribute—has become a regulatory expectation for biosimilar characterization [21] [23]. For size variants, SEC is typically complemented by analytical ultracentrifugation (AUC) and field-flow fractionation techniques to provide a comprehensive size variant profile across different size ranges [21].
SEC Applications Across the Biopharmaceutical Workflow
Process Development and Optimization
During bioprocess development, SEC serves as a vital monitoring tool for evaluating cell culture conditions, purification parameters, and formulation stability. It enables researchers to track undesirable aggregation or fragmentation that may occur during various manufacturing steps, allowing for rapid process optimization [18]. Real-time SEC data facilitates informed decision-making regarding harvest timing, purification column loading, and buffer exchange procedures, ultimately ensuring consistent product quality [23]. The implementation of high-throughput SEC methods further accelerates this development cycle by enabling parallel assessment of multiple process conditions.
Quality Control and Stability Testing
In quality control environments, validated SEC methods are routinely employed for lot release testing and stability monitoring of biopharmaceutical products [20] [22]. These methods quantitatively measure the distribution of monomeric protein relative to high-molecular-weight aggregates and low-molecular-weight fragments, all of which represent CQAs with potential implications for product safety and efficacy [22]. As required by regulatory guidelines, SEC methods for QC applications must undergo rigorous validation demonstrating specificity, accuracy, precision, linearity, and robustness according to ICH Q2(R1) guidelines [20] [22].
Table 1: SEC Validation Parameters for Quality Control Applications
| Validation Parameter | Experimental Approach | Acceptance Criteria |
|---|---|---|
| Specificity | Resolution of monomer, aggregate, and fragment peaks | Baseline separation (R > 1.5) between critical peak pairs |
| Accuracy/Recovery | Spiked samples with known concentrations | 95-105% recovery of monomeric protein |
| Precision | Repeatability (n=6) of same sample | RSD ≤ 2.0% for monomer percentage |
| Linearity | Series of standards across concentration range | R² ≥ 0.995 |
| Range | Concentrations demonstrating acceptable linearity, precision, accuracy | Typically 0.1-5 mg/mL for proteins |
| Robustness | Deliberate variations in flow rate, temperature, mobile phase | Consistent elution profile and monomer quantification |
Analytical Similarity Assessment of Biosimilars
SEC plays a foundational role in the comparative analytical assessment required for biosimilar development [21] [24]. According to the FDA's "totality-of-the-evidence" approach, comprehensive analytical characterization forms the base of the evidence pyramid for demonstrating biosimilarity [24]. SEC data on size variants contributes directly to establishing that a proposed biosimilar falls within the quality range established using multiple lots (typically 10-12) of the reference product [22]. This statistical approach captures the natural variability of the reference product while setting meaningful acceptance criteria for the biosimilar candidate [22].
Experimental Data and Case Studies
SEC Method Validation for Bevacizumab Biosimilar Development
A detailed SEC method validation study for bevacizumab analysis demonstrates the rigorous approach required in biosimilar assessment [22]. Researchers employed a Waters apparatus with a Protein KW-804 column and refractive index detection, using phosphate-buffered saline (300 mM NaCl, 25 mM phosphate, pH 7.0) as the mobile phase at 1.0 mL/min flow rate [22]. The method demonstrated excellent linearity (R² > 0.995) across the concentration range of 5-30 μg/mL, with precision showing RSD ≤ 0.35% for repeated injections [22]. The application of this validated method to multiple bevacizumab lots highlighted the importance of accounting for both analytical method variability and between-lot variability when establishing quality ranges for biosimilar assessment [22].
SEC Column Performance Comparison for Gene Therapy Products
A systematic evaluation of wide-pore SEC columns for characterizing gene therapy products revealed important performance considerations [19]. When analyzing rAAV serotypes, columns with pore sizes of 550-700 Å demonstrated optimal selectivity, though resolution was highly dependent on the specific serotype [19]. The DNACore AAV-SEC column showed particularly high efficiency (11,000 plates), attributed to its monodisperse 3 μm silica particles [19]. For mRNA analysis (1000-5000 nucleotides), the Biozen dSEC-7 LC column (700 Å) achieved the highest efficiency for smaller mRNA (~1000 nucleotides), while larger pore sizes were more appropriate for longer mRNA transcripts [19]. Despite these advancements, the study noted limitations in resolving low and high molecular weight species of mRNA across all tested columns, highlighting an area for continued technical development [19].
Table 2: SEC Column Performance for Different Biopharmaceutical Modalities
| Product Modality | Optimal Pore Size | Highest Performing Column | Key Separation Challenge |
|---|---|---|---|
| Monoclonal Antibodies | 150-300 Å | Not specified in results | Resolving low-abundance aggregates from main monomer peak |
| rAAV Serotypes | 550-700 Å | DNACore AAV-SEC (11,000 plates) | Selectivity highly dependent on specific serotype |
| mRNA (≤1000 nts) | ~700 Å | Biozen dSEC-7 LC | Limited resolution of LMWS and HMWS |
| mRNA (>1000 nts) | >700 Å | Columns with larger pore sizes | Accurate quantification of impurities |
Experimental Protocols and Workflows
Standard Operating Procedure for SEC Aggregate Analysis
Method: This protocol describes the quantitative determination of high-molecular-weight aggregates in therapeutic monoclonal antibodies using SEC with UV detection.
Materials and Equipment:
- HPLC system with UV detection capability
- SEC column appropriate for mAb analysis (e.g., 150-300 Å pore size)
- Mobile phase: Phosphate-buffered saline (300 mM NaCl, 25 mM phosphate, pH 7.0)
- Protein reference standard
- Sample filtration unit (0.22 μm)
Procedure:
- Mobile Phase Preparation: Prepare phosphate-buffered saline (300 mM NaCl, 25 mM phosphate, pH 7.0), filter through 0.22 μm filter, and degas.
- System Equilibration: Equilibrate the SEC column with mobile phase at 1.0 mL/min until stable baseline is achieved (typically 30-60 minutes).
- System Suitability Test: Inject protein reference standard to confirm resolution (R > 1.5 between monomer and aggregate peaks), tailing factor (<2.0), and column efficiency (>5,000 plates/m).
- Sample Preparation: Dilute protein sample to 1-2 mg/mL in mobile phase, centrifuge at 10,000 × g for 5 minutes, and transfer supernatant to HPLC vial.
- Chromatographic Analysis: Inject 25-50 μL of prepared sample, run isocratic elution at 1.0 mL/min for 30 minutes with UV detection at 280 nm.
- Data Analysis: Integrate peaks corresponding to high-molecular-weight species, monomer, and low-molecular-weight species. Calculate percentage of each species based on relative peak area.
Validation Parameters: Establish specificity, linearity (1-5 mg/mL), precision (RSD < 2%), accuracy (90-110% recovery), and limit of quantitation (0.1% for aggregates) [22].
Native SEC-MS Workflow for ADC Characterization
Method: This protocol describes the characterization of drug-to-antibody ratio (DAR) and drug load distribution in cysteine-linked antibody-drug conjugates using native SEC-MS.
Materials and Equipment:
- LC-MS system compatible with high molecular weight analysis
- SEC column with appropriate pore size for mAb separation
- Ammonium acetate buffer (100-200 mM, pH 6.5-7.5)
- ADC reference standard
Procedure:
- Mobile Phase Preparation: Prepare 200 mM ammonium acetate, pH 7.0, using LC-MS grade water and acetic acid. Filter through 0.22 μm filter and degas.
- MS Parameter Optimization: Set ESI source to low fragmentation conditions (cone voltage 80-120 V, source temperature 100-150°C) to maintain non-covalent complexes.
- Chromatographic Conditions: Isocratic elution with 200 mM ammonium acetate at 0.2-0.3 mL/min for 5-10 minutes.
- Sample Preparation: Dilute ADC to 5-10 μg/mL in ammonium acetate buffer, centrifuge at 10,000 × g for 5 minutes.
- Data Acquisition: Inject 5-10 μL of sample, monitor UV at 280 nm, and acquire MS data in positive ion mode with extended m/z range (500-4000 m/z).
- Data Analysis: Deconvolute mass spectra to determine average DAR and drug load distribution using vendor software [20].
Figure 1: Native SEC-MS Workflow for ADC Characterization
Essential Research Reagent Solutions
Table 3: Key Research Reagents and Materials for SEC Analysis
| Reagent/Material | Function/Purpose | Application Notes |
|---|---|---|
| SEC Columns | Separation based on hydrodynamic size | Pore size selection critical: 150-300 Å for mAbs, 550-700 Å for rAAVs, 700-1000 Å for mRNA [19] |
| Mobile Phase Buffers | Maintain protein integrity during separation | Phosphate-buffered saline common; ammonium acetate for native MS compatibility [20] [22] |
| Molecular Weight Standards | Column calibration and method validation | Protein standards for biotherapeutics; polynucleotides for gene therapy products |
| Reference Standards | System suitability testing | Well-characterized biologics for qualifying method performance |
| Sample Preparation Kits | Buffer exchange and desalting | Essential for transferring samples into compatible SEC mobile phase |
Regulatory Considerations and Quality Metrics
SEC in Biosimilarity Assessment
Global regulatory agencies, including the FDA and EMA, emphasize the importance of comprehensive analytical comparison in biosimilar development [21] [24]. The FDA recommends a "stepwise" approach where analytical assessment forms the foundation, potentially reducing the need for extensive clinical studies if sufficient analytical similarity is demonstrated [24]. SEC data on size variants contributes significantly to this assessment, particularly when employing the quality range (QR) method that compares biosimilar product attributes against the natural variability of the reference product established using 10-12 lots [22]. This approach requires thorough method validation and understanding of both analytical method variability and between-lot variability of the reference product [22].
Quantitative Metrics for SEC Separation Quality
Assessing SEC separation quality requires appropriate metrics beyond traditional resolution calculations, particularly when dealing with asymmetric peaks or significant differences in peak abundance [25]. The peak-to-valley ratio is often more applicable than resolution for SEC separations where the abundance of high and low molecular weight species is much lower than the main monomer peak [25]. Recent research proposes a Separation Quality Factor (QS) incorporating five normalized terms: separation of peak pairs, peak elution window, separation impedance, peak symmetry, and signal-to-noise ratio [25]. This comprehensive metric enables more objective comparison of different SEC columns and conditions, supporting robust method development [25].
Figure 2: SEC Quality Metrics for Regulatory Compliance
Size-exclusion chromatography maintains a critical position within the biopharmaceutical analytical toolbox, providing essential data on size variants throughout the product lifecycle. As biotherapeutic modalities expand to include ADCs, gene therapies, and other complex molecules, SEC methodologies continue to evolve through advanced detection systems, improved column chemistries, and integration with complementary techniques like mass spectrometry. The demonstrated applications across process development, quality control, and biosimilar assessment underscore SEC's fundamental role in ensuring product quality, safety, and efficacy. For biosimilar developers particularly, robust SEC methods generating high-quality comparative data form the analytical foundation supporting regulatory submissions under the totality-of-the-evidence approach. Continued refinement of SEC technologies and application-specific methodologies will further strengthen its contribution to biopharmaceutical development and quality assurance.
Integrating Self-Produced Sensors into Robust SEC Methods
Size-exclusion chromatography (SEC) is an indispensable analytical technique for the separation and characterization of macromolecules based on their hydrodynamic volume. The strategic selection of SEC columns—dictated by pore size, particle size, and column dimensions—is fundamental to achieving optimal resolution for specific applications. For researchers validating self-produced sensors, the choice of SEC column directly impacts the accuracy and reliability of characterizing sensor materials or biological conjugates. The separation mechanism relies on the differential access of molecules to the porous network of the stationary phase; larger molecules elute first as they are excluded from pores, while smaller molecules penetrate pores and elute later. A profound understanding of how column parameters influence this separation is crucial for effective method development.
Recent research underscores that no single SEC column consistently provides the best separation across all sample types [19]. The performance is highly sample-dependent, necessitating a systematic approach to column selection. This guide objectively compares current SEC column technologies and provides supporting experimental data to inform researchers, scientists, and drug development professionals in making strategic choices that enhance resolution, efficiency, and data validity for their specific needs, including sensor validation workflows.
Core Column Parameters and Their Impact on Resolution
The resolution in SEC is governed by three primary physical parameters of the column: pore size, particle size, and the physical dimensions of the column itself. Each parameter interacts with the sample to determine the final separation quality.
Pore Size: The pore size of the stationary phase particles determines the molecular weight (MW) separation range. Molecules separate effectively when their size falls within the fractionation range of the pores. For instance, in gene therapy product characterization, optimal selectivity for recombinant adeno-associated viruses (rAAVs) was found with columns having larger pore sizes of 550–700 Å, whereas for larger mRNAs (>1000 nucleotides), columns with even larger pore sizes (up to 1000 Å) were more appropriate [19]. Selecting a pore size matched to the hydrodynamic volume of the analytes is the first critical step.
Particle Size: The particle size of the packing material directly influences column efficiency and backpressure. Smaller particles generally provide higher efficiency (more theoretical plates) and better resolution but also generate higher system backpressure, requiring instrumentation capable of handling such pressures. A recent study comparing SEC columns for gene therapy products found that a column with monodisperse 3 µm silica particles achieved a high efficiency of 11,000 theoretical plates, whereas a column with 5 µm particles showed significantly lower efficiency (< 1000 plates) [19]. Advances in column technology have led to the proliferation of sub-2 µm particles for UHPLC-SEC, enabling faster and higher-resolution analyses.
Column Dimensions: The length and internal diameter (ID) of a column impact resolution, analysis time, and sample loading capacity. A longer column generally provides higher resolution but increases analysis time and backpressure. Shorter columns enable faster analysis, ideal for high-throughput environments. Sample loading is typically limited to a maximum of 1% of the total column volume [26]. For analytical applications, common dimensions include 7.8 mm ID x 30 cm L, with a typical sample load of 15-150 µL of a 1-10 mg/mL solution. For narrower 4.6 mm ID columns, the load decreases to 1-10 µL of a similar concentration [26].
Table 1: Effect of Core SEC Column Parameters on Separation
| Parameter | Primary Impact | Effect on Resolution | Practical Consideration |
|---|---|---|---|
| Pore Size | Determines MW separation range | Highest when analyte size is within the fractionation range | Match pore size to analyte's hydrodynamic volume; not all pore sizes work for all analytes [19] [26]. |
| Particle Size | Influences column efficiency (theoretical plates) | Smaller particles generally increase resolution | Balance higher resolution (smaller particles) with system pressure capabilities [19]. |
| Column Length | Impacts theoretical plates and analysis time | Longer columns increase resolution | Weigh need for resolution against longer run times and higher backpressure [26]. |
| Column Internal Diameter (ID) | Affects sample loading capacity | Minimal direct effect, but overloading causes poor resolution | Do not exceed 1% of total column volume; scale load with column ID [26]. |
Comparative Performance Data for Different Applications
The performance of SEC columns varies significantly across different application domains, such as biologics, gene therapies, and synthetic polymers. Below, we summarize key experimental findings from a systematic comparison of wide-pore SEC columns for characterizing gene therapy products, which provides a model for objective evaluation.
A 2025 study published in the Journal of Chromatography A systematically evaluated a new generation of wide-pore SEC columns for analyzing messenger RNA (mRNA) and recombinant adeno-associated viruses (rAAVs) [19]. The research offers critical, data-driven insights for column selection.
For rAAV Serotypes: Among six tested SEC columns with pore sizes from 450 to 700 Å, the DNACore AAV-SEC column, packed with monodisperse 3 µm silica particles, demonstrated the highest efficiency with approximately 11,000 theoretical plates. This was attributed to its small, uniform particle size. In contrast, the SRT SEC-500 column, with 5 µm particles, showed the lowest efficiency (< 1000 plates). The study concluded that columns with larger pore sizes (550–700 Å) provided optimal rAAV selectivity. However, a crucial finding was that the final resolution for different rAAV serotypes was highly sample-dependent, with no single column consistently providing the best separation across all variants [19].
For mRNA Analysis: The study evaluated five columns with pore sizes from 700 to 1000 Å. The Biozen dSEC-7 LC (700 Å) column systematically achieved the highest efficiency and was particularly well-suited for analyzing smaller mRNA of around 1000 nucleotides. For larger mRNAs exceeding 1000 nucleotides, columns with larger pore sizes were more appropriate. A significant limitation noted across all tested columns was the challenge in accurately separating and quantifying low and high molecular weight species (LMWS and HMWS) of mRNA, which affects integration and quantification accuracy [19].
Table 2: SEC Column Performance in Gene Therapy Product Characterization [19]
| Analyte | Optimal Pore Size | High-Performing Column Example | Key Performance Metric | Limitations Noted |
|---|---|---|---|---|
| rAAV Serotypes | 550 - 700 Å | DNACore AAV-SEC | ~11,000 theoretical plates | Resolution highly sample-dependent; no single column was universally best. |
| Small mRNA (~1000 nts) | ~700 Å | Biozen dSEC-7 LC | Systematically highest efficiency | Accurate separation and quantification of LMWS/HMWS remained limited. |
| Large mRNA (>1000 nts) | >700 Å (up to 1000 Å) | Columns with larger pore sizes | More appropriate for size | Accurate separation and quantification of LMWS/HMWS remained limited. |
Experimental Protocols for SEC Column Evaluation
Adopting a standardized experimental approach is vital for objectively comparing SEC columns and validating their performance for a specific application, such as characterizing sensor-biomolecule conjugates. The following protocols outline key methodologies for system testing and accuracy validation.
Protocol 1: System Performance Testing with Protein Standards
This protocol assesses the basic performance and efficiency of an SEC system and column using a characterized protein standard mixture.
- Principle: A mixture of well-defined proteins of different molecular weights is injected onto the SEC column. The elution profile is used to calculate the number of theoretical plates, a key indicator of column efficiency [26].
- Materials:
- SEC System: HPLC or UHPLC system with UV detection.
- Mobile Phase: Aqueous buffer, e.g., 100 mM phosphate buffer with 100 mM Na₂SO₄ or NaCl, pH 6.8 [26].
- Protein Standard: Commercially available SEC protein standard mixture (e.g., Merck/Sigma SEC Protein Standard 15–600 kDa or BioRad Protein Standard) [26].
- Procedure:
- Equilibrate the column with at least 5 column volumes of mobile phase at the intended flow rate.
- Prepare the protein standard according to the manufacturer's instructions.
- Inject the recommended volume (e.g., 15-150 µL for a 7.8 mm ID column) [26].
- Run the separation method and record the chromatogram.
- Identify a well-resolved, late-eluting peak (e.g., vitamin B12 or p-aminobenzoic acid) for plate count calculation.
- Calculate the number of theoretical plates (N) using the formula specified for your region (US, EU, Japan differ), where N = 16*(tᵣ/W)², tᵣ is the retention time of the peak, and W is the peak width at the baseline [26].
Protocol 2: Accuracy Validation Using Polydisperse Standards
This advanced procedure, adapted from established validation methodologies, determines the accuracy of an SEC method for determining molecular weight distributions [7].
- Principle: A polydisperse reference standard is prepared by mixing two monodisperse standards with certified molecular weights. The accuracy of the SEC method is calculated by comparing the experimental molecular weight averages (Mₙ and M_w) against the known "true" values of the prepared mixture [7].
- Materials:
- Two monodisperse polymer or protein standards with known molecular weights (M₁ and M₂).
- Analytical balance, appropriate solvents, and volumetric flasks.
- Procedure:
- Generate an SEC calibration curve using primary or secondary monodisperse standards.
- Analyze representative samples to obtain average Mₙ and Mw values.
- Calculate the required M₁ and M₂ for a two-component mixture that matches the sample's Mₙ and Mw using the formulas for an equal-weight mixture [7]:
- Mₙ = 2 / (1/M₁ + 1/M₂)
- Mw = (M₁ + M₂) / 2
- Weigh equal amounts of the two selected monodisperse standards to create the validation mixture.
- Analyze the prepared standard in triplicate using the SEC method.
- Determine the experimental (Mₙ)ₑₓₚ and (Mw)ₑₓₚ.
- Calculate the percent accuracy (relative error) for Mₙ and M_w [7]:
- % (Mₙ)ᴿᴱ = |(Mₙ)ₜ - (Mₙ)ₑₓₚ| / (Mₙ)ₜ * 100%
- % (Mw)ᴿᴱ = |(Mw)ₜ - (Mw)ₑₓₚ| / (Mw)ₜ * 100% where subscript 't' denotes the true (calculated) value.
Strategic Selection Workflow and Critical Considerations
Integrating the parameters and data, the following workflow provides a logical, stepwise strategy for selecting an SEC column to achieve optimal resolution. This process is summarized in the diagram below.
Diagram 1: A strategic workflow for selecting an SEC column to achieve optimal resolution. The process begins with analyte and solvent identification and proceeds through critical choices of matrix, pore size, and physical parameters before final method validation.
Key Decision Factors in the Workflow
Analyte & Solvent → Base Matrix: The chemical nature of the analyte and the required solvent dictate the base matrix of the stationary phase. Silica-based columns (e.g., TSKgel SW-type) are robust and efficient but may require specific mobile phases to minimize secondary interactions with the silica surface. They can often tolerate a percentage of organic solvent. Polymer-based columns (e.g., TSKgel PW-type) are more inert, offering superior compatibility with alkaline pH conditions, making them suitable for applications like oligonucleotide analysis [26].
Pore Size Selection: The molecular weight or size of the analyte is the primary guide for pore size selection. Vendors provide molecular weight ranges for their columns. It is critical to remember that SEC separates by hydrodynamic volume, not molecular weight. Therefore, the calibration curve should be constructed using standards that are structurally similar to the analyte (e.g., use protein standards for proteins, dextrans for polysaccharides) for accurate molecular weight estimation [26].
Mobile Phase Optimization: The mobile phase in SEC is not merely a carrier; it is crucial for suppressing unwanted secondary interactions (electrostatic, hydrophobic) between the analyte and the stationary phase. To minimize these interactions and achieve a pure size-based separation, use a buffer with an ionic strength ≥ 200 mM (e.g., 100 mM phosphate buffer plus 100 mM Na₂SO₄) [26]. For silica-based columns, conditioning with successive injections of a protein like BSA (e.g., 4x 100 µg) can saturate active sites and improve protein recovery [26].
Essential Research Reagent Solutions
A well-equipped SEC laboratory requires more than just a column. The following reagents and materials are essential for method development, system calibration, and routine performance testing.
Table 3: Key Research Reagents and Materials for SEC Analysis
| Reagent/Material | Function/Purpose | Application Example |
|---|---|---|
| Protein Standards Mix | System performance testing and calibration | Merck/Sigma SEC Protein Standard (15-600 kDa) for calculating theoretical plates and calibrating for protein analysis [26]. |
| Monodisp Polymer Stds | Accuracy validation of MW determination | Narrow dispersity polymers (e.g., PEGs) used to create polydisperse standards for validating SEC method accuracy per published protocols [7]. |
| BSA (Bovine Serum Albumin) | Column conditioning | Successive injections of BSA (e.g., 4x 100 µg) saturate active sites on silica-based SEC columns, improving recovery of analytical proteins [26]. |
| High-Purity Buffers & Salts | Mobile phase preparation | Phosphate buffers with Na₂SO₄ or NaCl (~200 mM total ionic strength) suppress secondary interactions for a pure SEC separation [26]. |
| Pore Size Calibration Kits | Column characterization and selection | Sets of standards with known molecular weights and sizes used to establish the calibration curve and fractionation range of a new column [26]. |
The strategic selection of SEC columns is a multidimensional process that balances pore size, particle size, and column dimensions against the specific characteristics of the target analyte and the requirements of the analytical method. As demonstrated by recent comparative studies, performance is highly application-specific, necessitating an informed and often empirical approach to selection [19]. For researchers validating self-produced sensors, a rigorous, protocol-driven evaluation of SEC columns—assessing efficiency, resolution, and accuracy—is paramount. By leveraging the workflows, performance data, and experimental protocols outlined in this guide, scientists can make objective, data-backed decisions that ensure optimal SEC resolution, thereby enhancing the reliability and credibility of their characterization data.
Size-exclusion chromatography (SEC) is a powerful technique for the separation and characterization of macromolecules based on their hydrodynamic volume. However, the ideal performance of SEC relies on a pure size-based separation mechanism. The presence of non-size effects, such as hydrophobic or ionic interactions between the analyte and the stationary phase, can severely compromise the accuracy of molecular weight determination. These aberrant interactions lead to skewed retention times, altered elution volumes, and ultimately, inaccurate molecular weight data. Consequently, the meticulous optimization of the mobile phase is not merely an improvement but a fundamental requirement for generating valid, reproducible results in SEC. For researchers validating self-produced sensors, where data fidelity is paramount, controlling these parameters is critical. This guide provides a systematic, data-driven approach to mobile phase optimization, balancing ionic strength, pH, and buffer composition to suppress non-size effects and achieve true size-based separations.
Fundamentals of Non-Size Effects in SEC
In an ideal SEC separation, molecules traverse the column packed with a porous medium, and their retention is governed solely by their ability to access the pore volume. Larger molecules that cannot enter the pores elute first, while smaller molecules that can penetrate the pores are retained longer. This process creates a calibration curve where the logarithm of molecular weight is inversely proportional to the elution volume [27].
Non-size effects disrupt this ideal behavior. The two most common types are:
- Hydrophobic Interactions: Occur between non-polar regions of the analyte and the stationary phase, leading to increased retention beyond what is expected based on size.
- Ionic Interactions: Occur between charged groups on the analyte and ionic sites on the stationary phase. These can cause either unwanted retention (attraction) or exclusion (repulsion), distorting the chromatogram.
The primary goal of mobile phase optimization is to create an environment that effectively masks these non-size effects, allowing the inherent size-exclusion mechanism to dominate the separation process.
Core Mobile Phase Parameters and Optimization Strategies
The composition of the mobile phase is the most critical tool for mitigating non-size effects. The following table summarizes the key parameters, their impact on separation, and optimization targets.
Table 1: Key Mobile Phase Parameters for Minimizing Non-Size Effects
| Parameter | Primary Function | Effect on Non-Size Interactions | Typical Optimization Range |
|---|---|---|---|
| Ionic Strength | Modulates electrostatic interactions between analyte and stationary phase. | High ionic strength shields charges, suppressing ionic interactions. | 0.1 - 0.5 M for salts like NaCl [28] [7] |
| pH | Controls the charge state of the analyte and stationary phase surface. | Adjusting pH away from the analyte's isoelectric point (pI) can increase charge repulsion; matching pH can minimize attractive forces. | pH 6.0 - 8.0 for many biomolecules; specific to analyte pI [28] |
| Buffer Composition | Provides buffering capacity to maintain stable pH and can directly influence interactions. | Choice of buffer ions (e.g., phosphate, Tris, ammonium acetate) can affect specific binding and MS-compatibility. | 10 - 100 mM for adequate buffering capacity [28] |
| Organic Modifiers | Modifies the polarity of the mobile phase. | Low percentages (e.g., 1-5%) of acetonitrile or methanol can disrupt hydrophobic interactions. | < 5% to avoid altering stationary phase or denaturing proteins |
Optimizing Ionic Strength
Ionic strength is paramount for suppressing electrostatic non-size effects. The dissolved salts in the mobile phase provide counterions that shield electrostatic attractions between the analyte and the stationary phase.
- Experimental Protocol for Ionic Strength Titration:
- Prepare a series of mobile phases with a constant pH and buffer concentration but varying concentrations of a neutral salt like sodium chloride (e.g., 0, 0.1, 0.2, 0.3 M).
- Inject a standard protein or polymer mixture with known molecular weights and varied pIs.
- Monitor the elution volume of each standard. As ionic strength increases, the elution volumes of analytes affected by ionic interactions will shift.
- The optimal ionic strength is the lowest concentration at which elution volumes become constant and align with the expected molecular weight calibration curve [28] [7].
Optimizing pH
The pH of the mobile phase determines the net charge of proteins and the ionization state of silica-based stationary phases.
- Experimental Protocol for pH Scouting:
- Prepare mobile phases with a constant ionic strength and buffer concentration across a relevant pH range (e.g., pH 5.0, 6.0, 7.0, 8.0).
- Use buffers with good buffering capacity in the target range (e.g., phosphate for 6-8, acetate for 4-5.5).
- Inject the standard mixture and analyze the chromatograms for peak shape, symmetry, and retention time.
- The optimal pH is often one where the analyte and stationary surface carry the same charge, leading to electrostatic repulsion rather than attraction, or where the analyte is highly charged to minimize hydrophobic interactions [28].
Balancing Buffer Composition and Additives
The buffer system must maintain a stable pH while being compatible with the analytes and detection system (e.g., mass spectrometry).
- Volatile Buffers for MS-Compatibility: For hyphenation with mass spectrometry, volatile salts like ammonium acetate or ammonium bicarbonate are essential. Research on ion-exchange chromatography of monoclonal antibodies has shown that a mixture of 50 mM ammonium acetate and 50 mM ammonium carbonate can provide sufficient ionic strength and buffering capacity while being fully MS-compatible [28].
- Additives for Hydrophobicity: For analytes with significant hydrophobic character, adding 2-5% methanol or acetonitrile can improve peak shape and recovery. The use of additives like 0.1% trifluoroacetic acid (TFA) for proteins or amines for basic compounds is common in other chromatographic modes and can be carefully evaluated in SEC, though they may influence the stationary phase [29].
Experimental Data and Comparative Analysis
The following table synthesizes experimental data from published studies to illustrate the impact of mobile phase conditions on key SEC performance metrics.
Table 2: Comparative Experimental Data on Mobile Phase Optimization
| Analyte | Mobile Phase Condition A | Mobile Phase Condition B | Observed Effect (A vs. B) | Key Takeaway |
|---|---|---|---|---|
| Monoclonal Antibodies [28] | 10-40 mM Ammonium Acetate/Carbonate (Low Ionic Strength) | 50/50 mM Ammonium Acetate/Carbonate (≥70 mM Ionic Strength) | Condition A: Poor peak shape, variable retention. Condition B: Improved peak shape, stable retention, good MS signal. | Total ionic strength should be >70 mM for proper elution and peak shape of proteins. |
| Polydisperse Polymer Standards [7] | Sub-optimal ionic strength/pH | Optimized ionic strength/pH | Accurate determination of Mn and Mw requires mobile phases that suppress non-size effects for validation. | Mobile phase optimization is critical for accuracy validation in SEC. |
| Pharmaceutical/Cosmetic Compounds [30] | Various SFC mobile phases on Cyanopropyl (CN) column | Same compounds on other stationary phases | The CN stationary phase in SFC showed the best correlation between retention and skin permeability, highlighting the role of stationary phase selection. | The stationary phase itself is a key variable in controlling interactions. |
A Workflow for Systematic Mobile Phase Optimization
The following diagram maps the logical sequence of experiments for a structured approach to mobile phase optimization.
The Scientist's Toolkit: Essential Research Reagents
The following table lists key reagents and materials required for the experimental protocols described in this guide.
Table 3: Essential Reagents and Materials for SEC Mobile Phase Optimization
| Reagent/Material | Function | Example Use Case |
|---|---|---|
| High-Purity Salts (NaCl, KCl, (NH₄)₂CO₃) | Adjusts ionic strength to suppress electrostatic interactions. | General purpose SEC for proteins and polymers. |
| MS-Compatible Volatile Salts (Ammonium Acetate, Formate) | Provides ionic strength for MS detection; avoids ion source contamination. | SEC-MS hyphenation for protein therapeutic characterization [28]. |
| Buffering Agents (Phosphate, Tris, Acetate) | Maintains stable pH throughout the separation. | Controlling charge state of analytes and stationary phase. |
| HPLC-Grade Organic Modifiers (Acetonitrile, Methanol) | Disrupts hydrophobic interactions between analyte and stationary phase. | Improving peak shape and recovery of hydrophobic proteins/peptides. |
| Narrow-Disperse Molecular Weight Standards | Calibrates the SEC system and validates the suppression of non-size effects. | Accuracy validation of the final SEC method [7] [27]. |
The validation of self-produced sensors and the acquisition of reliable macromolecular characterization data in SEC are inextricably linked to the mastery of mobile phase optimization. Non-size effects pose a significant threat to data integrity, but they can be systematically identified and suppressed. By understanding the roles of ionic strength, pH, and buffer composition, and by employing a structured experimental workflow, researchers can achieve robust, reproducible SEC methods. The experimental data and protocols provided here serve as a foundation for developing mobile phases that ensure separations are governed by size alone, thereby solidifying the credibility of subsequent scientific conclusions.
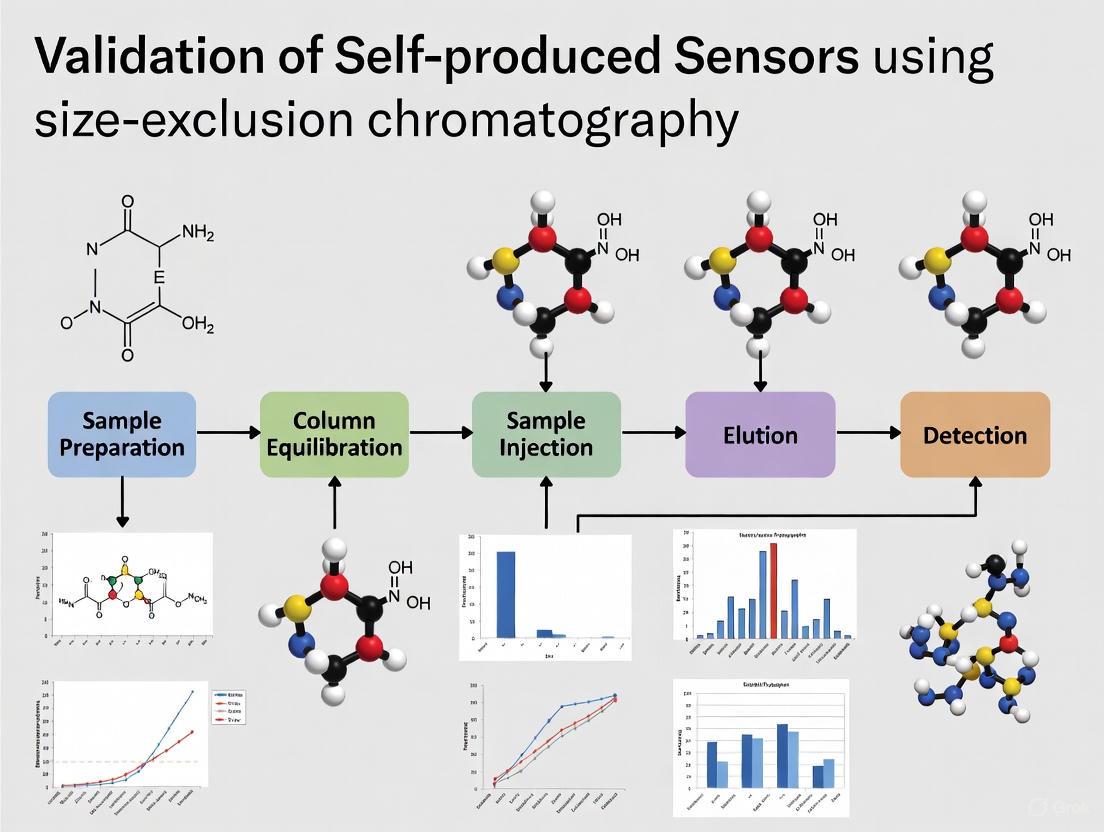
Sensor integration is a foundational process in modern scientific laboratories, particularly for researchers and scientists in drug development who rely on precise, reliable data. The process involves combining various sensors into a unified system to gather, share, and analyze environmental data, enabling intelligent decision-making [31]. Within the specific context of validating self-produced sensors—such as those for monitoring critical parameters in Size-Exclusion Chromatography (SEC) systems—a structured protocol ensures that the collected data is accurate, reproducible, and fit-for-purpose. This guide provides a step-by-step protocol for sensor integration, from physical connection to data acquisition, and objectively compares the performance of different sensor integration approaches with supporting experimental data.
Section 1: Foundational Concepts of Sensor Integration
What is Sensor Integration?
Sensor integration is the process of combining different types of sensors into a single, cohesive system. This allows devices to collect comprehensive information from the environment, analyze it, and make autonomous decisions based on the synthesized data. In an SEC lab setting, this could involve integrating temperature, pressure, and flow sensors to monitor and control chromatographic conditions in real-time [31].
The Critical Role of Sensor Performance Metrics
When integrating sensors, especially for validation purposes, understanding key performance metrics is essential. These metrics allow for the quantitative comparison of sensor quality and are vital for validating self-produced sensors against reference instruments.
- Accuracy: Refers to how close a sensor's measurement is to the true or target value. It is assessed by comparing the sensor's readings with those from a reference-grade instrument [32].
- Precision: Describes the consistency and repeatability of measurements. A precise sensor will produce similar results when the same quantity is measured multiple times, even if those results are not perfectly accurate [32].
- Reproducibility: A specific aspect of precision, reproducibility is determined by placing multiple sensors of the same kind in one location and observing how much their measurements differ from one another [32].
A clear example of these concepts is sensor drift, where a sensor's measurements progressively deviate from the reference over time. This indicates declining accuracy, even if the sensor maintains high precision and reproducibility. Such drift can often be corrected with compensation algorithms, highlighting the importance of ongoing validation [32].
Section 2: A Step-by-Step Sensor Integration Protocol
Step 1: Sensor Selection and Compatibility Check
The first step is choosing the correct sensor for your application.
- Define the Use Case: Clearly identify what needs to be measured (e.g., temperature, pressure, vibration) and the environmental conditions it will operate in [33].
- Match Sensor to Application: For an SEC lab, you might need temperature sensors for column ovens or pressure sensors for pump monitoring. Ensure the sensor's accuracy, resolution, response time, and durability meet the experimental requirements. Industrial-grade sensors are often necessary for harsh environments [33].
- Verify Hardware & Software Compatibility: Ensure the sensor is compatible with your existing gateways, programmable logic controllers (PLCs), or microcontrollers. Check communication interfaces and protocols from the outset [33].
Step 2: Establishing Physical and Communication Connections
The method of connecting the sensor to your data system depends on the application's needs for speed, complexity, and distance.
- Direct Connection: This method involves a direct link between the sensor and a microcontroller (e.g., via I2C, SPI, UART). It is ideal for applications demanding rapid responses, such as triggering an immediate alarm or control action [34].
- Gateway-Based Integration: In this scenario, multiple sensors connect to a central gateway device. The gateway collects and processes data before transmitting it to a cloud or central system. This approach is excellent for managing diverse sensors from various manufacturers and is well-suited for complex SEC setups where sensors are scattered [34].
- Cloud-Based Integration: Here, sensor data is sent to the cloud for storage and heavy processing. This can happen directly from the sensor or via a gateway. It enables extensive data analysis and remote access but relies on a stable internet connection [34].
- Hybrid Integration: This approach balances local and cloud processing. An edge device (like a gateway) performs initial data analysis on-site, filtering out critical information. For instance, it might send an alert to the cloud only if a temperature spike is detected, enabling real-time local responses while leveraging the cloud for deeper analysis [34].
Step 3: Power Management and Physical Installation
- Prioritize Power Efficiency: For remote or battery-powered setups, opt for low-power sensors with sleep modes and fast wake-up cycles. Evaluate the total power budget and sensor duty cycles carefully [33].
- Rugged Installation: Consider the physical location and mounting requirements. In harsh laboratory environments, sensors may need rugged enclosures and weatherproofing to perform as intended. Proper installation by skilled professionals is often required for accurate measurements [33].
Step 4: Data Acquisition, Processing, and Security
- Establish Data Requirements: Define the type of data (digital, analog), frequency of collection, and data volume. This drives the selection of storage and processing solutions [33].
- Implement Data Processing:
- Device-Level: Basic processing on the sensor itself for quick responses.
- Edge Processing: More complex tasks on a nearby gateway, combining data from multiple devices and reducing bandwidth use by sending only essential information upstream [34].
- Cloud Processing: Heavy analysis, complex computations, and machine learning applied to large datasets from multiple sources [34].
- Implement Security Measures: Cybersecurity is critical. Use sensors that support data encryption, authentication, and secure firmware updates to protect sensitive experimental data [33].
Section 3: Validation and Performance Comparison for Self-Produced Sensors
Experimental Protocol for Sensor Validation
Validating a self-produced sensor involves a direct comparison against a reference instrument under controlled conditions.
- Co-location Testing: Place the sensor under test and a reference-grade instrument in the same location to measure the same parameter simultaneously [32].
- Controlled Environment Testing: For the highest precision, place the sensor for a long time in a controlled environment (e.g., a test chamber) with a defined stimulus to see how much its readings vary [32].
- Data Collection & Analysis: Collect data from both the test sensor and the reference instrument. Use performance parameters like the coefficient of determination (R²) and Root Mean Square Error (RMSE) to quantify the agreement between them [32].
Performance Comparison: Sensor Quality vs. Data Acquisition Strategy
A critical trade-off in sensor integration exists between sensor quality and data acquisition strategy. Research has evaluated how sensor quality (level of noise) and data storage (monitoring interval) impact the accuracy of prognostic systems, such as predicting a system's Remaining Useful Life (RUL).
The table below summarizes findings from a trade-off analysis, showing how different combinations of sensor quality and data intervals affect prognostic performance [35].
Table 1: Trade-off Analysis between Sensor Quality and Data Interval on Prognosis Performance
| Data Interval (Cycle) | Noise Level (Uniform Distribution) | Impact on RUL Prediction Accuracy |
|---|---|---|
| Small (Δt=1) | Small (0.2) | High accuracy and low uncertainty |
| Small (Δt=1) | Large (0.5) | Reduced accuracy due to high noise |
| Large (Δt=8) | Small (0.2) | Reduced accuracy due to sparse data |
| Large (Δt=8) | Large (0.5) | Lowest accuracy and high uncertainty |
Key Insight: The study concluded that using high-quality sensors (low noise) is highly effective. However, if high-quality sensors are unavailable, increasing the frequency of data measurements (reducing the data interval) can compensate and improve prediction accuracy, even when using lower-quality, more noisy sensors [35]. This finding is crucial for labs validating self-produced sensors, which may initially have higher noise levels.
Workflow Diagram: Sensor Integration and Validation
The following diagram illustrates the complete workflow for integrating and validating a sensor, from selection to performance assessment.
Section 4: The Scientist's Toolkit for Sensor Integration
Table 2: Essential Research Reagent Solutions for Sensor Integration and SEC Validation
| Item | Function in Sensor Integration / SEC Validation |
|---|---|
| Microcontroller (e.g., Arduino, Raspberry Pi) | Acts as a central processing unit for directly connected sensors; receives and processes raw data [31]. |
| IoT Gateway | A hub device that collects data from diverse sensors using different protocols, enables local (edge) processing, and transmits data to the cloud [34]. |
| Communication Protocol Stacks (e.g., MQTT, CoAP, Zigbee) | Software libraries that enable the sensor to communicate data efficiently over the chosen protocol, ensuring reliable transmission [33] [34]. |
| Reference-Grade Instrument | An officially recognized, high-precision instrument used as a scientific standard to validate the accuracy of the self-produced sensor [32]. |
| Certified Reference Materials (CRMs) | Well-characterized materials used to calibrate sensors and assess the trueness and precision of measurements during validation [7]. |
| Size-Exclusion Chromatography (SEC) Columns | Used in the validation thesis to fractionate polydisperse polymers like β-glucans by hydrodynamic radius, providing molecular weight distribution data [36]. |
| Triple-Detector Array (TDA) for SEC | A system comprising a light scatterer, a viscometer, and a refractive-index detector. It provides comprehensive characterization of molecular parameters (MW, intrinsic viscosity) for analytes, serving as a rich data source for sensor validation [36]. |
Section 5: Advanced Considerations for a Modern Lab
The Role of AI and Automation
The laboratory landscape is rapidly evolving with automation and AI. Instrumentation is increasingly capable of autonomous optimization. For example, machine learning can be used to autonomously refine liquid chromatography gradients, improving reproducibility and data quality while reducing manual input [37]. These technologies can be leveraged to create more robust and self-correcting sensor systems.
Ensuring Scalability and Long-Term Value
- Plan for Scalability: Choose sensors and architectures that allow for the easy addition of new sensors as research needs grow. Using standardized protocols simplifies future expansion [33].
- Optimize Cost vs. Performance: Balance sensor quality and features with the total cost of ownership, including installation, maintenance, and energy costs, not just the initial purchase price [33].
- Work with Trusted Vendors: Partner with reliable sensor manufacturers and system integrators to ensure access to documentation, technical support, and long-term product availability [33].
A meticulous, step-by-step approach to sensor integration—from careful selection and connection to rigorous validation—is paramount for generating reliable data in scientific research. For drug development professionals validating self-produced sensors within SEC research, this protocol provides a clear roadmap. The experimental data demonstrates that a strategic trade-off between sensor quality and data acquisition frequency can be leveraged to achieve reliable performance. By adhering to these guidelines and utilizing the provided toolkit, researchers can build a solid foundation for their sensor-integrated systems, ensuring data accuracy that meets the stringent demands of modern laboratories.
Size-exclusion chromatography (SEC) coupled with multiple detection systems provides a powerful platform for comprehensive biomolecular characterization, particularly in the validation of novel sensors and analytical methods. The integration of Ultra-Violet (UV), Multi-Angle Light Scattering (MALS), Refractive Index (RI), and Fluorescence detectors creates an analytical system where each detector contributes unique but complementary information about molecular properties. This multi-attribute monitoring approach is especially valuable in biopharmaceutical development, where characterizing critical quality attributes (CQAs) of complex molecules like monoclonal antibodies (mAbs) and antibody-drug conjugates (ADCs) is essential for ensuring product safety and efficacy [38] [39]. For researchers validating self-produced sensors, this detector combination offers a robust framework for cross-validation and method verification, providing absolute measurements of molar mass, size, concentration, and conformational changes without reliance on reference standards.
Detector Principles and Complementary Data
Fundamental Operating Principles
Each detector in the SEC-UV-MALS-RI-Fluorescence array operates on different physical principles to yield specific macromolecular parameters:
- UV Detection: Measures absorbance of ultraviolet light by chromophores in the sample (e.g., protein aromatic amino acids at 280 nm). Provides concentration information and identifies peaks based on spectral characteristics [40].
- Multi-Angle Light Scattering (MALS): Measures the intensity of scattered light at multiple angles to determine absolute molar mass and size (radius of gyration) without column calibration [41]. Governed by the relationship between measured light scattering intensity (Rθ) and molar mass (M).
- Refractive Index (RI) Detection: Measures the change in refractive index between the mobile phase and the solute. Provides concentration information independent of chromophore presence and is particularly valuable for molecules with poor UV absorbance [40].
- Fluorescence Detection: Measures emitted light following excitation at specific wavelengths. Offers exceptional sensitivity and selectivity for fluorescent molecules or those tagged with fluorophores [41].
Data Complementarity in Multi-Detector Systems
The power of this detection strategy lies in the complementary nature of the information generated. While UV and RI both provide concentration measurements, their agreement validates accuracy, while discrepancies can reveal matrix effects or chromophore abnormalities. MALS provides absolute molar mass that is independent of elution position, correcting for potential inaccurate molecular weight estimations from SEC alone. Fluorescence adds another dimension of specificity, enabling tracking of particular fluorescent species within complex mixtures. When all detectors are aligned temporally, a comprehensive profile emerges for each eluting fraction, providing molar mass, size, concentration, and structural integrity simultaneously.
Experimental Comparison of Detection Strategies
Performance Metrics for Detector Configurations
Table 1: Quantitative comparison of detector performance in SEC analysis of monoclonal antibodies
| Detection Method | Molar Mass Accuracy (%) | Aggregate Detection Sensitivity (ng) | Concentration Range (mg/mL) | Fluorescence Interference Impact |
|---|---|---|---|---|
| SEC-UV (280 nm) | ±15-20 (relative) | 50-100 | 0.01-10 | Minimal effect |
| SEC-MALS | ±2-5 (absolute) | 10-50 | 0.05-5 | Significant over-estimation [41] |
| SEC-RI | ±10-15 (relative) | 100-200 | 0.1-20 | Negligible effect |
| SEC-Fluorescence | N/A | 1-10 (for fluorescent species) | 0.001-1 | Target-specific |
| SEC-MALS with FIC | ±3-5 (absolute) | 10-50 | 0.05-5 | Corrected over-estimation [41] |
Case Study: Fluorescence Interference Correction in MALS Measurements
A critical challenge in combining these detection methods is fluorescence interference in light scattering measurements. Fluorescent macromolecules can introduce tremendous error in MALS determinations, leading to significant over-estimation of molar mass when fluoresced photons are measured as scattered photons [41]. Standard mitigation involves installing bandwidth filters (typically 10-20 nm) to suppress detection of fluorescent emissions. However, research demonstrates that for strongly fluorescing systems like IR800CW-conjugated proteins or intrinsically fluorescent lignins, these filters alone are insufficient [41].
The Fluorescence Interference Correction (FIC) procedure developed for MALS quantifies and corrects for residual fluorescence transmission. The methodology calculates the transmission factor (Tf) representing the fraction of fluorescence that passes through bandwidth filters, enabling mathematical extraction of the pure scattering component (RθS) using the equation:
RθS = (Rθ,filtered/(1-Tf)) - (Tf × Rθ,unfiltered/(1-Tf))
where Rθ,filtered and Rθ,unfiltered represent the Raleigh ratios from filtered and unfiltered detectors, respectively [41]. This correction enables accurate molar mass determination of fluorescent macromolecules that would otherwise yield erroneous results, expanding MALS applicability to fluorescent proteins, fluorescence-tagged proteins, and optically active nanoparticles.
Validation of Multi-Attribute Methods
The combination of SEC with multiple detectors aligns with the pharmaceutical industry's move toward Multi-Attribute Methods (MAM) that use mass spectrometry to monitor multiple product quality attributes simultaneously [38] [39]. While HRAM MS provides the foundation for MAM, the physical characterization provided by SEC-UV-MALS-RI-Fluorescence offers complementary data on size variants and aggregates that may not be detectable by MS alone. Studies validating SEC methods for monoclonal antibody aggregate analysis have demonstrated that light scattering provides superior sensitivity for aggregate detection compared to UV alone, as aggregates scatter light more efficiently than they absorb UV light [40].
Experimental Protocols for Method Validation
Protocol for SEC-UV-RI-MALS Analysis of Protein Aggregates
Materials and System Configuration:
- Chromatography System: HPLC or UHPLC system (e.g., Shimadzu LC-20A)
- SEC Column: TSKgel G3000SWxl, 5 µm, 7.8 mm i.d. × 30 cm L
- Mobile Phase: PBS, filtered through 0.1 µm membrane
- Flow Rate: 1 mL/min
- Injection Volume: 20 µL
- Detection: UV (280 nm), RI, and MALS (e.g., miniDAWN TREOS) detectors in series [40]
Critical Method Parameters:
- Maintain system cleanliness with regular flushing (1% SDS for 4-5 hours, followed by water and 20% ethanol)
- Use 0.05% sodium azide in aqueous buffers to prevent bacterial growth
- Filter all buffers through 0.1 µm membrane to reduce baseline noise
- Ensure proper degassing of eluents, particularly for organic solvents
- Perform system suitability tests with standard samples before analysis sequences
- Use RI signal as concentration source when UV-absorbing additives are present [40]
Protocol for Fluorescence Interference Correction in MALS
Materials:
- MALS instrument with multiple detectors (e.g., Wyatt Technology instruments)
- Bandwidth filters (typically ±10 nm) installed before MALS detectors
- Fluorescent standards (e.g., IR800CW-conjugated Bovine Serum Albumin)
- Fluorimetry capability for pre-determination of emission spectra [41]
Procedure:
- Prior to MALS experimentation, obtain emission spectrum of fluorescent sample using standard fluorimetry
- Calculate transmission rates of fluorescence through bandwidth filters based on emission characteristics
- Perform SEC-MALS analysis with both filtered and unfiltered detectors
- For each measurement angle, match filtered detectors with nearest-angle unfiltered detectors
- Apply FIC procedure using predetermined Tf values to calculate pure scattering component RθS
- Use corrected RθS values for molar mass calculations [41]
Validation Approach: Validate the FIC procedure using proteins with known conjugation ratios of fluorophores. Compare molar mass results before and after correction with theoretical values. For non-fluorescent standards, filtered and unfiltered detectors should yield identical results, confirming proper system alignment.
Workflow Visualization: Multi-Detector SEC Analysis
Figure 1: Integrated SEC Multi-Detector Workflow with Fluorescence Correction
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key reagents and materials for SEC multi-detector analysis
| Research Reagent | Function in Analysis | Application Notes |
|---|---|---|
| TSKgel SWxl SEC Columns | High-resolution size-based separation | Suitable for proteins; clean with 0.1M Glycine/HCl pH 3 [40] |
| IR800CW Fluorophore | Fluorescent tagging for detection | Used in FIC validation; narrow emission simplifies correction [41] |
| Immobilized Trypsin Kits | Protein digestion for MAM | Enables reproducible peptide mapping; essential for HRAM MS [38] |
| PBS with 0.05% Sodium Azide | Aqueous mobile phase | Prevents bacterial growth; change buffers without azide daily [40] |
| Bovine Serum Albumin | Protein standard for validation | Used as conjugate base for fluorescence correction studies [41] |
| SMART Digest Kits | Standardized protein digestion | Provides high reproducibility for peptide mapping; automation compatible [38] |
| Bandwidth Filters (±10 nm) | Fluorescence suppression in MALS | Blocks red-shifted fluoresced light; standard in MALS systems [41] |
Data Integration and Analysis Strategies
Unified Data Platforms for Multi-Detector Systems
A significant challenge in implementing advanced detection strategies is managing and integrating data from multiple instrument sources. Chromatography data systems must harmonize information from UV, MALS, RI, and fluorescence detectors, each potentially operating with different sampling rates and data formats. Centralized data analysis platforms improve accuracy and reproducibility by applying uniform processing parameters (integration algorithms, calibration models) across all detector channels [42]. These platforms create controlled environments where detector responses can be temporally aligned and cross-correlated, enabling comprehensive molecular characterization that transcends the capabilities of individual detectors.
Advanced Applications: From Characterization to QC
The transition of multi-detector SEC from characterization to quality control environments represents the evolution of these techniques toward routine application. Pre-study and in-study validation of SEC methods with different detection modes follows ICH-Q2(R1) guidelines, employing control charts and quality control samples to monitor process parameters [43]. For biopharmaceutical analysis, MAM approaches that combine peptide mapping with high-resolution mass spectrometry are increasingly adopted for monitoring multiple product quality attributes simultaneously [38] [39]. The physical characterization provided by SEC-UV-MALS-RI-Fluorescence complements these MS-based methods by providing information on higher-order structure and aggregation that may not be evident at the peptide level.
Logical Framework for Detection Selection
Figure 2: Decision Framework for Detector Selection and Application
The strategic combination of UV, MALS, RI, and Fluorescence detection in SEC analysis creates a powerful platform for multi-attribute monitoring that exceeds the capabilities of any single detection method. This approach is particularly valuable for researchers validating self-produced sensors, as it provides multiple orthogonal measurement principles for cross-validation. The development of correction methods for technical challenges such as fluorescence interference in MALS measurements further enhances the accuracy and applicability of these techniques. As the biopharmaceutical industry advances toward more comprehensive characterization strategies, the integration of physical characterization methods with emerging technologies like MAM and HRAM MS will provide unprecedented insight into product quality attributes, ultimately leading to safer and more effective biotherapeutics.
The accurate characterization of complex biological samples, such as adeno-associated viruses (AAVs) and monoclonal antibodies (mAbs), is a critical requirement in biopharmaceutical development and quality control. These innovative therapeutics present unique analytical challenges due to their structural complexity and heterogeneity. Size-exclusion chromatography (SEC) has emerged as a powerful orthogonal technique for assessing critical quality attributes (CQAs) including size variants, aggregates, and capsid content [44] [45]. Within the broader context of validating self-produced sensors for chromatographic research, understanding the performance characteristics of different analytical approaches becomes paramount. This comparison guide objectively evaluates current SEC methodologies and competing technologies for characterizing AAVs and mAbs, providing experimental data and protocols to inform analytical strategy decisions for researchers, scientists, and drug development professionals.
The fundamental challenge in analyzing AAVs and mAbs lies in their inherent heterogeneity. AAV samples typically contain mixtures of empty capsids, partially filled capsids, and fully assembled vectors alongside process-related impurities [46] [47]. Similarly, mAbs exhibit glycoform heterogeneity that significantly impacts their effector function, safety, and pharmacokinetics [48]. This guide systematically compares the performance of various analytical techniques to address these characterization challenges, with particular emphasis on method capabilities, limitations, and implementation requirements.
Comparative Analysis of Techniques for AAV and mAb Characterization
Size-exclusion chromatography (SEC) separates biomolecules based on their hydrodynamic volume as they pass through a column packed with porous particles [44] [45]. For AAV analysis, SEC effectively resolves empty and full capsids while providing information on aggregates and fragments [44]. When coupled with multi-angle light scattering (MALS) detection, SEC enables precise determination of molar mass and size [45] [47]. The technique's primary advantages include high throughput, excellent reproducibility, and ease of implementation in quality control environments [46]. However, SEC-MALS has limitations in resolving capsids with intermediate DNA content and requires careful method optimization to avoid secondary interactions [45] [47].
Analytical ultracentrifugation (AUC) utilizes centrifugal force to separate particles based on their buoyant density [47]. In sedimentation velocity mode (AUC-SV), empty capsids (approximately 66S) and full capsids (approximately 105S) can be resolved based on their distinct sedimentation coefficients [47]. AUC is considered a gold standard technique for capsid content quantification due to its high resolution and ability to detect intermediate species [47]. The method directly measures particle properties without reference standards, but requires specialized equipment, extensive expertise, and has relatively low throughput [46] [47].
Charge detection mass spectrometry (CDMS) is a single-particle technique that simultaneously measures mass-to-charge ratio (m/z) and charge (z) for individual ions [47]. Empty capsids (approximately 3.8 MDa) and full capsids (approximately 5.3 MDa) appear as distinct peaks in CDMS spectra, enabling precise quantification [47]. This label-free method provides excellent mass accuracy and can detect heterogeneous populations, but requires advanced instrumentation and specialized data analysis expertise [46] [47].
Mass photometry measures the mass of individual particles by detecting the light scattered as they pass through a glass slide [46]. The technique requires minimal sample preparation, consumes only small sample volumes, and provides results within minutes [46]. Mass photometry can distinguish empty, partially filled, and full capsids based on mass differences, but has limitations in determining absolute titer [46].
Table 1: Comparison of Key Analytical Techniques for AAV Characterization
| Technique | Measurement Principle | Key Applications | Throughput | Resolution | Implementation Complexity |
|---|---|---|---|---|---|
| SEC-MALS | Hydrodynamic size separation with light scattering detection | Empty/full ratio, aggregates, size variants | High | Moderate | Moderate |
| AUC | Sedimentation velocity in centrifugal field | Empty/full/intermediate capsid quantification | Low | High | High |
| CDMS | Single-particle mass measurement | Empty/full/intermediate capsid mass determination | Moderate | High | High |
| Mass Photometry | Light scattering of individual particles | Empty/full ratio, mass distribution | High | Moderate | Low |
| cryo-EM | Electron microscopy with classification | Visualization and counting of capsid species | Low | High | High |
Quantitative Performance Comparison
A comprehensive study comparing orthogonal techniques for AAV capsid content quantification revealed significant methodological differences [47]. Researchers prepared spike samples with defined ratios of empty and full capsids and analyzed them using multiple technologies. The results demonstrated that AUC most closely matched the theoretical values across the entire range of spike ratios, confirming its status as a reference method [47]. CDMS also showed excellent agreement with expected values, particularly for samples with 0-50% empty capsids [47].
Table 2: Quantitative Comparison of Empty Capsid Percentage Across Analytical Techniques [47]
| Theoretical % Empty | % Empty by AUC | % Empty by CDMS | % Empty by cryo-EM | % Empty by SEC-MALS | SEC A260/A280 |
|---|---|---|---|---|---|
| 100% | 100% | 100% | 99% | 100% | 0.58 |
| 83% | 85% | 84% | 95% | 90% | 0.87 |
| 75% | 76% | 78% | 92% | 81% | 0.97 |
| 50% | 55% | 49% | 74% | 58% | 1.17 |
| 33% | 41% | 38% | 67% | 40% | 1.27 |
| 20% | 23% | 18% | 51% | 24% | 1.32 |
| 0% | 3% | 0% | 14% | 0% | 1.39 |
Notably, cryo-EM consistently overestimated empty capsid content across all samples, potentially due to classification challenges or imaging artifacts [47]. SEC-MALS performed well at extreme ratios but showed some deviation at intermediate values (33-50% empty), possibly due to limited resolution of partially filled capsids [47]. The A260/A280 ratio trended predictably with capsid content, providing a rapid assessment method though with lower resolution than orthogonal techniques [47].
For mAb characterization, supercritical fluid chromatography-tandem mass spectrometry (SFC-MS/MS) has demonstrated remarkable performance for glycoform analysis [48]. This platform achieved detection of 102 glycoforms across five therapeutic mAbs (bevacizumab, nivolumab, ramucirumab, rituximab, and trastuzumab) in just 8 minutes, with a detection limit of 5 attomoles and a dynamic range exceeding 6 orders of magnitude [48]. This represents a significant advancement over conventional fluorescence HPLC analysis, which typically offers only 3 orders of magnitude dynamic range [48].
Experimental Protocols for Key Characterization Methods
SEC Method Development for AAV Analysis
Column Selection Protocol: Systematic column evaluation is essential for optimal SEC method development. A recent study compared six wide-pore SEC columns, finding that no single column performed best for all AAV samples [44]. Key considerations include:
- Pore Size Matching: Select columns with appropriate pore size matched to the target AAV size using the Peak Positioning Term (Q4) metric [44]. Columns such as BiozenSEC-7 LC, AdvanceBio SEC 500A, and SurePac Bio 550 SEC MDi have demonstrated high Q4 scores for AAV samples [44].
- Particle Characteristics: Monodisperse silica columns (e.g., SurePac Bio 550 SEC MDi) provide higher efficiency compared to polydisperse columns due to more uniform packing [44].
- Column Hardware: Use inert column hardware to minimize protein adsorption and improve recovery [44].
- Mobile Phase Optimization: Employ low-flow rates to enhance resolution between aggregates and monomers, even with short UHPLC columns [44].
Sample Preparation and Analysis: To preserve sample integrity, use low-binding glass vials instead of plastic to prevent AAV adsorption [44]. For SEC-MALS analysis, inject larger AAV amounts to maintain resolution while accommodating MALS detection sensitivity requirements [44]. When developing methods for multiple AAV serotypes, note that SEC-MALS is serotype-agnostic as it relies on physical parameters rather than molecular recognition [46].
AUC Method for Capsid Content Quantification
Sample Analysis Protocol: AUC sedimentation velocity analysis enables direct quantification of empty, intermediate, and full AAV capsids [47]:
- Data Collection: Acquire both A260 and A280 absorbance scans during centrifugation. Empty capsids sediment at approximately 66S while full capsids sediment at approximately 105S [47].
- Peak Identification: Identify species based on sedimentation coefficients and characteristic A260/A280 ratios: approximately 0.59 for empty capsids and 1.39 for full capsids [47].
- Quantification: Use interference data for quantitative analysis of species distribution since the refractive index increment (dn/dc) shows minimal variation (approximately 2%) between empty (dn/dc ≈ 0.185) and full (dn/dc ≈ 0.181) capsids [47].
- Data Processing: Integrate peak areas for individual species in the c(s) distribution plots to determine relative percentages of empty, intermediate, and full capsids [47].
SFC-MS/MS Method for mAb Glycoform Analysis
Glycan Release and Derivatization: Release N-glycans from mAbs using Rapid PNGase F according to manufacturer protocols [48]. Extract glycans using Sepharose CL-4B beads with 85% acetonitrile conditioning [48]. Derivatize released glycans through peracetylation with pyridine and acetic anhydride at 50°C for 4 hours [48].
Chromatographic Separation: Perform SFC-MS analysis using a supercritical fluid chromatography system with the following parameters:
- Column: Shim-pack UC-Phenyl (2.1 × 150 mm, 3 μm)
- Mobile Phase: CO2 with methanol modifier
- Gradient: 10% to 40% B over 4 minutes, hold at 40% B for 3 minutes
- Make-up Solvent: Methanol with 0.1% ammonium formate at 0.1 mL/min
- Detection: Triple-quadrupole mass spectrometer with MRM mode monitoring m/z 210 fragment ion [48]
Data Analysis: Process acquired data using appropriate software, utilizing the product ion chromatogram area for m/z 210 for glycoform quantification [48]. Calculate percentage composition of each glycoform relative to total glycan content [48].
Workflow Visualization of Analytical Approaches
Figure 1: Analytical Workflow Selection for AAV and mAb Characterization
Research Reagent Solutions for AAV and mAb Characterization
Table 3: Essential Research Reagents and Materials for AAV and mAb Analysis
| Reagent/Material | Function/Application | Key Characteristics | Example Usage |
|---|---|---|---|
| Wide-pore SEC Columns | Separation of AAV size variants | Appropriate pore size (e.g., 500Å), monodisperse particles, inert hardware | Resolving empty and full AAV capsids [44] [45] |
| Sepharose CL-4B Beads | Glycan extraction and purification | Porous carbohydrate matrix for size exclusion | Purifying released N-glycans from mAbs [48] |
| Rapid PNGase F | Enzymatic release of N-glycans | High efficiency and rapid cleavage | Releasing glycans from monoclonal antibodies for analysis [48] |
| Low-binding Glass Vials | Sample storage and preparation | Minimal surface adsorption | Preserving AAV recovery during storage and analysis [44] |
| Peracetylation Reagents | Glycan derivatization | Improves MS detection sensitivity | Preparing mAb glycans for SFC-MS analysis [48] |
| MALS Detector | Absolute molecular weight determination | Multi-angle light scattering measurement | Coupling with SEC for AAV size and mass analysis [45] [47] |
Implications for Sensor Validation in Chromatographic Research
The comparative performance data and experimental protocols presented herein provide a critical framework for validating self-produced sensors in SEC research. When developing novel sensing technologies for chromatographic applications, researchers must consider several key factors emerging from this analysis:
Reference Standard Correlation: New sensor technologies should demonstrate strong correlation with orthogonal methods, particularly for challenging applications like intermediate capsid detection where techniques vary significantly in performance [47]. The discrepancies observed between AUC, CDMS, cryo-EM, and SEC-MALS highlight the importance of using multiple reference methods during validation [47].
Resolution and Sensitivity Requirements: Sensor capabilities must align with analytical needs, such as the requirement to detect low-abundance glycoforms in mAbs (attomole sensitivity) [48] or resolve partially filled AAV capsids [46] [47]. The 6-order magnitude dynamic range demonstrated by SFC-MS/MS for glycoform analysis sets a challenging benchmark for novel sensor development [48].
Serotype-Agnostic Performance: For AAV analysis, sensors should maintain performance across different serotypes without requiring extensive re-optimization, similar to physical characterization methods like SEC-MALS, AUC, and mass photometry [46].
Throughput and Implementation Balance: Ideal sensor technologies balance the high resolution of reference methods (e.g., AUC, CDMS) with the practical implementation advantages of techniques like SEC-MALS and mass photometry [46] [47]. The validation framework should assess both analytical performance and practical utility in regulated environments.
This comparison guide provides a foundation for evaluating both established and emerging analytical technologies, enabling researchers to make informed decisions when developing and validating novel sensor platforms for characterizing complex biologics.
Solving Common SEC Challenges for Reliable Sensor Data
Size-exclusion chromatography (SEC) is a foundational technique for the analysis of proteins and other biomacromolecules, separating analytes based on their hydrodynamic volume or molecular weight [49]. The ideal SEC mechanism is a purely entropic process, where separation is governed by the differential partitioning of molecules between the mobile phase and the pores of the stationary phase, without any adsorptive interactions. However, non-size exclusion effects, particularly hydrophobic and ionic interactions between the analyte and the stationary phase, frequently complicate this ideal behavior. These undesirable interactions can lead to inaccurate molecular weight determinations, reduced recovery, poor resolution, and peak tailing, ultimately compromising the reliability of analytical results [50] [49].
For researchers engaged in the validation of self-produced sensors, such as the self-validating (SEVA) sensors described in the literature, understanding and controlling these effects is paramount [51]. The validation of a novel sensor often requires precise characterization of its components, including proteins used as recognition elements. The presence of non-size exclusion effects during SEC analysis can yield misleading data on the purity, aggregation state, or molecular weight of these biomolecules, thereby invalidating the sensor's calibration and performance metrics. This guide provides a comparative analysis of methodologies to identify and mitigate these confounding interactions, ensuring the integrity of SEC data within a sensor development workflow.
Background and Fundamental Concepts
Self-Validating Sensors and Analytical Validation
The concept of self-validating sensors represents a significant advancement in measurement reliability. As outlined by Henry et al., a SEVA sensor is an intelligent integrated system that goes beyond simple measurement to perform online monitoring of its own status [51] [52]. It outputs not only a measurement value but also key parameters such as a "confirmed measurement value" and an associated "measurement uncertainty value," providing a real-time indication of data quality [51]. The development and validation of such sophisticated sensors often rely on foundational analytical techniques like SEC to characterize biological components. Consequently, the accuracy of the SEC data directly influences the validated performance of the sensor itself.
Mechanisms of Non-Size Exclusion Effects
In practice, the stationary phases used in SEC are not perfectly inert. Two primary types of interactions can occur:
- Hydrophobic Interactions: These occur between non-polar regions on the analyte and hydrophobic sites on the stationary phase. The strength of these interactions is influenced by the ionic strength and composition of the mobile phase [50] [53]. At high ionic strengths, the "salting-out" effect can enhance hydrophobic interactions, leading to increased retention beyond what is expected based on size alone [53].
- Ionic (Electrostatic) Interactions: These occur between charged groups on the analyte and oppositely charged functional groups on the stationary phase. These interactions can either attract an analyte to the surface (increasing retention) or repel it (decreasing retention), and are highly dependent on the pH and ionic strength of the mobile phase [50].
Table 1: Characteristics of Non-Size Exclusion Interactions in SEC.
| Interaction Type | Molecular Driving Force | Impact on Analyte Retention | Key Influencing Factors |
|---|---|---|---|
| Hydrophobic | Association of non-polar surfaces in an aqueous environment | Increases retention time | High ionic strength, mobile phase organic content, temperature |
| Ionic (Attractive) | Electrostatic attraction between oppositely charged groups | Increases retention time | pH, low ionic strength |
| Ionic (Repulsive) | Electrostatic repulsion between similarly charged groups | Decreases retention time (can lead to elution in void volume) | pH, low ionic strength |
Figure 1: Mechanisms and impacts of non-size exclusion effects in SEC. Hydrophobic interactions are strengthened by high ionic strength, while ionic interactions are typically weakened.
Experimental Protocols for Identifying Interactions
A systematic approach is required to diagnose the presence and type of non-size exclusion effects interfering with a separation.
Systematic Mobile Phase Screening
The most direct method for identifying interactions is to observe how analyte retention changes as a function of mobile phase composition.
Protocol for Ionic Interaction Assessment:
- Prepare a series of mobile phase buffers with a constant pH but varying ionic strength (e.g., 0.05 M, 0.1 M, 0.2 M sodium phosphate, all at pH 7.0).
- Inject the protein sample of interest onto the SEC column equilibrated with each buffer.
- Plot the retention time or elution volume of the analyte against the ionic strength. A significant trend of decreasing retention time with increasing ionic strength is a clear indicator that attractive ionic interactions are occurring and are being shielded by the salt.
Protocol for Hydrophobic Interaction Assessment:
- Prepare mobile phases with a constant, moderately high ionic strength (e.g., 0.2 - 0.5 M NaCl) to suppress ionic effects.
- Incorporate small, controlled percentages of a modifier known to disrupt hydrophobic interactions (e.g., 2-10% ethanol, isopropanol, or acetonitrile) or a non-ionic detergent.
- Inject the sample. A notable decrease in retention time with the addition of organic modifier confirms the presence of hydrophobic interactions.
Quantitative Assessment of Separation Quality
When optimizing SEC methods to mitigate these effects, it is crucial to move beyond simple retention time observations and use quantitative descriptors of separation quality. Fekete and Imiolek propose a Separation Quality Factor (QS) that integrates multiple parameters into a single, normalized metric [25]. This is particularly useful when comparing the performance of different columns or mobile phase conditions.
The QS factor is calculated from five normalized terms (Q₁ to Q₅):
- Q₁ (Peak Pair Separation): Uses a normalized peak-to-valley (p/v) ratio, which is more robust than resolution (Rₛ) for SEC peaks of vastly different heights.
- Q₂ (Number of Peaks): Rewards separations that resolve a greater number of distinct peaks.
- Q₃ (Peak Distribution): Favours a uniform distribution of peaks across the chromatographic window.
- Q₄ (Analysis Time): Penalizes excessively long analysis times.
- Q₅ (Peak Symmetry): Rewards symmetric, non-tailed peaks.
The overall QS is the product of these terms, providing a comprehensive metric to guide method development away from conditions that promote undesirable interactions, which often manifest as peak tailing and poor resolution [25].
Comparative Data and Mitigation Strategies
Once non-size exclusion effects are identified, selecting the appropriate mitigation strategy is essential. The following table compares the primary approaches, synthesized from the reviewed literature.
Table 2: Comparison of Strategies to Mitigate Non-Size Exclusion Effects in SEC.
| Mitigation Strategy | Mechanism of Action | Optimal Use Case | Key Limitations & Considerations | Supporting Experimental Data |
|---|---|---|---|---|
| Mobile Phase Ionic Strength Adjustment | Shields electrostatic charges, disrupting ionic interactions. | When ionic interactions are suspected or confirmed. | Excessively high ionic strength can promote hydrophobic interactions ("salting-out") [53]. | A study on protein LLPS showed electrostatic forces are screened out at high salt (>1.5 M), eliminating ionic effects [53]. |
| Mobile Phase pH Modification | Alters the net charge of the analyte and stationary phase surface. | When the analyte's isoelectric point (pI) is known. Can be used to introduce repulsive forces. | pH must be compatible with analyte stability and column operating range. | Chromatography literature indicates that undesirable ionic interactions at low ionic strength can be mitigated by adjusting pH [50]. |
| Organic Modifiers (e.g., <10% ACN, MeOH) | Reduces the hydrophobic effect by decreasing solvent polarity. | When hydrophobic interactions are the primary issue. | Can denature some proteins; not always compatible with all SEC stationary phases (e.g., silica-based). | The addition of modifiers is a standard chromatographic practice to disrupt hydrophobic binding [50]. |
| Additives (e.g., Chaotropic Salts, Arginine) | Disrupts the structure of water, weakening the hydrophobic effect. | For stubborn hydrophobic interactions where organic modifiers fail or denature the protein. | May require extensive optimization for concentration; can be difficult to remove from the protein post-purification. | Used in protein chemistry to prevent aggregation and undesirable adsorption [50]. |
| Column Selection (e.g., High Salt Tolerance, Specialty Phases) | Physically separates analytes using a stationary phase with minimal residual charge or hydrophobicity. | For methods requiring high robustness or when analyzing a wide range of diverse analytes. | Can be more expensive than standard SEC columns. | Novel SEC device designs focus on improving resolution by optimizing flow dynamics, but surface chemistry remains critical [49]. |
The data in Table 2 demonstrates that there is no single universal solution. The optimal strategy often involves a balanced combination of these approaches. For instance, a method might employ a moderately high ionic strength (e.g., 0.3 - 0.5 M NaCl) to suppress ionic interactions, coupled with a small percentage of a compatible organic modifier (e.g., 2-5% isopropanol) to control any residual hydrophobic effects, all while operating at a pH that ensures protein stability and maximizes charge-based repulsion from the column matrix.
The Scientist's Toolkit: Essential Research Reagents
Successful identification and mitigation of non-size exclusion effects require a set of standard reagents and materials. The following table details key solutions and their functions in SEC method development.
Table 3: Key Research Reagent Solutions for SEC Troubleshooting.
| Reagent / Solution | Primary Function in SEC | Brief Rationale & Application Note |
|---|---|---|
| Sodium Phosphate Buffer | Provides buffering capacity and variable ionic strength. | The standard buffer for SEC; allows systematic study of ionic strength effects from 0.05 M to 0.5 M. |
| Sodium Chloride (NaCl) | Ionic strength adjuster. | Used to incrementally increase the ionic strength of the mobile phase to shield electrostatic interactions. |
| Potassium Chloride (KCl) | Ionic strength adjuster. | An alternative to NaCl; useful for comparing salt-specific effects, though NaCl is more common. |
| Ethanol or Isopropanol | Hydrophobic interaction disruptor. | Organic modifiers added at low percentages (2-10%) to disrupt hydrophobic interactions without denaturing most proteins. |
| Arginine Hydrochloride | Multi-functional additive. | Effective at suppressing both hydrophobic and ionic interactions, often used in protein formulation and chromatography at 0.1-0.5 M concentrations. |
| Urea or Guanidine HCl | Chaotropic agents. | Disrupts water structure and hydrophobic interactions; use with caution as they are denaturants. |
| Non-ionic Detergents (e.g., Tween-20) | Surfactant. | Coats hydrophobic surfaces, preventing adsorption; typically used at 0.01-0.05% concentration. |
Within the critical context of validating self-produced sensors, the integrity of analytical data used for component characterization is non-negotiable. Hydrophobic and ionic interactions represent significant sources of error in SEC, a technique often relied upon for this purpose. This guide has outlined a clear pathway for researchers: first, to systematically identify these non-size exclusion effects through controlled changes in mobile phase composition and quantitative assessment of chromatographic quality; and second, to effectively mitigate them using a comparative understanding of strategies ranging from mobile phase optimization to strategic column selection.
By adopting these practices, scientists can ensure that their SEC data accurately reflects the molecular properties of their sensor's biological elements, thereby strengthening the validation process for the entire SEVA system and ensuring the reliability of the final sensor's output. A rigorous, data-driven approach to SEC method development is not merely a chromatographic exercise, but a foundational element in the chain of validation for advanced sensor technologies.
Optimizing Flow Rate and Temperature for Enhanced Efficiency and Reproducibility
In the field of drug development and biologics analysis, Size-Exclusion Chromatography (SEC) is a critical technique for the separation and analysis of macromolecules based on their hydrodynamic volume [50]. For researchers, particularly those engaged in validating self-produced sensors, achieving high levels of efficiency and reproducibility is paramount. The optimization of operational parameters, most notably flow rate and temperature, is a fundamental prerequisite for obtaining reliable and consistent data [50]. This guide objectively compares the performance implications of different flow rate and temperature settings, providing supporting experimental data and detailed methodologies to inform analytical protocols.
Core Principles of SEC Optimization
The separation mechanism in SEC is purely based on the differential diffusion of molecules into and out of the pores of the stationary phase [50]. Unlike other chromatographic techniques, it does not rely on chemical interactions between the analyte and the stationary phase. This unique mechanism makes parameters like flow rate and temperature especially critical, as they directly influence this diffusion process and, consequently, the quality of the separation, the level of aggregation detected, and the reproducibility of results [50].
Comparative Analysis of Key Parameters
Flow Rate Optimization
The flow rate of the mobile phase is a dominant factor in SEC separations. Using a slow flow rate allows molecules sufficient time to diffuse into and out of the static pool of mobile phase contained within the pore structure, which is essential for achieving optimal separation [50].
Table 1: Impact of Flow Rate on SEC Performance
| Flow Rate (for 7.8 mm i.d. column) | Impact on Separation Efficiency | Impact on Analysis Time | Impact on Pressure | Recommended Use Case |
|---|---|---|---|---|
| ~1.0 mL/min (Standard) [50] | Good resolution for many applications | Baseline analysis time | Standard operating pressure | General quality control; established methods |
| < 1.0 mL/min (Slower) [50] | Higher resolution; sharper peaks; minimizes on-column broadening [54] | Increased | Lower | Complex mixtures; analyzing large proteins like antibodies [50] |
| > 1.0 mL/min (Faster) [50] | Reduced resolution; potential for inaccurate aggregation data | Decreased; higher throughput | Increased | Sample screening during early development; when resolution is sufficient [50] |
For larger molecules such as proteins, the optimum flow rate is typically much lower than for small molecules. For instance, while a small molecule might be analyzed at 1.2 mL/min on a 300 mm × 7.8 mm column, a protein may require a flow rate of 0.6 mL/min for optimal efficiency, significantly affecting run times [50]. When scaling to columns with different internal diameters, the flow rate should be adjusted to maintain a similar linear velocity. For a 4.6-mm i.d. column, the normal flow rate of 1.0 mL/min for a 7.8-mm i.d. column translates to approximately 0.35 mL/min [50].
Temperature Control and Optimization
Temperature is often overlooked in isocratic SEC methods, with many methods stating only "ambient" conditions. However, this can be a significant source of irreproducibility [50].
Table 2: Impact of Temperature on SEC Performance and Reproducibility
| Temperature Condition | Impact on Separation | Impact on Reproducibility & System | Risk of Aggregation |
|---|---|---|---|
| Ambient (Uncontrolled) [50] | Variable diffusion rates; changing peak shape and retention times | Low reproducibility; column pressure and viscosity change with room temperature fluctuations | Variable |
| Controlled (e.g., 25°C) [50] | Consistent diffusion and peak sharpness | High reproducibility; stable system pressure and viscosity | Controlled |
| Elevated (e.g., > 40°C) [50] | Faster diffusion can lead to sharper peaks and better resolution | Very stable viscosity and pressure | High risk; can cause sample precipitation |
A laboratory environment where the ambient temperature changes more than 10 °C can lead to a noticeable impact on the results. The change in temperature alters the viscosity of the mobile phase, which in turn affects the column operating pressure and the diffusion process into and out of the pore structure [50].
Experimental Protocols for Parameter Validation
Protocol for Determining Optimal Flow Rate
- Column Selection: Use a defined SEC column (e.g., 7.8 mm i.d. x 30 cm length) and a standardized mobile phase [50].
- Sample Preparation: Prepare a test mixture representative of your target analytes, such as a protein monomer and its known aggregates [50].
- Chromatographic Procedure: Inject the sample repeatedly, varying the flow rate across a range (e.g., 0.4 mL/min, 0.6 mL/min, 0.8 mL/min, 1.0 mL/min). Ensure other parameters (temperature, injection volume) are held constant [50].
- Data Analysis: Calculate the resolution (Rs) between critical peak pairs (e.g., monomer and dimer) and the theoretical plate height (H) for the main peak at each flow rate.
- Optimization: Plot H against the linear flow velocity (u). The minimum plate height (Hmin) corresponds to the optimum linear flow velocity (uopt), which provides the best separation efficiency [54]. The goal is to find a flow rate that provides the required resolution while maintaining an acceptable run time.
Protocol for Establishing Temperature Stability
- System Setup: Equip the SEC system with a column oven. Use the same column and a stable test sample as in the flow rate experiment [50].
- Chromatographic Procedure: Perform a series of injections at a fixed, optimized flow rate while systematically varying the column temperature (e.g., 15°C, 25°C, 35°C).
- Data Analysis: Measure the retention time and peak width of a key analyte (e.g., the monomer peak) across all injections.
- Reproducibility Assessment: Calculate the relative standard deviation (RSD%) of the retention times and peak widths for replicates at each temperature. A well-controlled temperature will yield a very low RSD% (e.g., <0.5%), demonstrating high reproducibility [50] [54].
- Aggregation Check: Monitor the chromatogram for the appearance of new, early-eluting peaks (indicating aggregates) at higher temperatures [50].
The diagram below illustrates the logical workflow for the systematic optimization of SEC methods.
Figure 1: SEC Method Optimization Workflow
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for SEC Analysis
| Item | Function in SEC Analysis |
|---|---|
| SEC Columns (Silica-based) [50] | The stationary phase that separates molecules based on their size. Pore size must be matched to the target analytes (a rule of thumb is 3x the molecule's diameter) [50]. |
| Monodisperse Polymer Standards [7] | Certified reference materials with known molecular weights used to generate the SEC calibration curve (log MW vs. elution volume) [7]. |
| Mobile Phase Buffers [50] | Aqueous solutions with defined ionic strength, pH, and composition. Essential for maintaining protein stability and preventing undesirable interactions with the stationary phase [50]. |
| Polydisperse Reference Standard [7] | A well-characterized mixture used for accuracy validation of the SEC method by comparing experimental molecular weight averages to known true values [7]. |
The optimization of flow rate and temperature is not merely a procedural step but a critical foundation for achieving efficient and reproducible results in Size-Exclusion Chromatography. A controlled, method-specific flow rate ensures optimal resolution and accurate aggregation detection, while precise temperature control is indispensable for mitigating run-to-run variability. For researchers validating self-produced sensors, adhering to the systematic experimental protocols and utilizing the essential tools outlined in this guide provides a robust framework for generating reliable, high-quality data that can withstand rigorous scientific scrutiny.
In the purification and analysis of therapeutic proteins, particularly monoclonal antibodies (mAbs), on-column aggregation presents a significant challenge that directly compromises sample recovery, analytical accuracy, and product quality. Unlike solution-based aggregation, this phenomenon occurs specifically during the chromatographic process when proteins undergo structural destabilization upon interaction with the stationary phase [55]. The resulting aggregates not only diminish yield but also pose serious clinical risks, as they can trigger undesirable immune responses in patients [55]. Understanding and addressing the mechanisms behind adsorption-induced aggregation is therefore crucial for developing robust purification protocols, especially in the context of validating novel analytical sensors where recovery and accuracy are paramount.
The following diagram illustrates the typical workflow for investigating on-column aggregation and the corresponding mitigation strategies, integrating both analytical separation and detection phases.
Experimental Evidence and Comparative Data
The phenomenon of on-column aggregation is well-documented in scientific literature, with several studies providing quantitative insights into its impact on sample recovery across different chromatographic modes.
Aggregation in Ion-Exchange Chromatography (IEX)
Ion-exchange chromatography (IEX), particularly cation-exchange (CEX), is widely used as a polishing step in mAb purification platforms due to its high loading capacity and selectivity for charge variants [55]. However, several studies have indicated the structural destabilization of various proteins upon adsorption on IEX resins. A mechanistic study on a model monoclonal antibody documented that adsorption on a strong CEX medium led to multi-peak elution behavior, a key indicator of protein unfolding and aggregation [55]. The study reported that the late-eluting peaks consisted of both native monomer and aggregates formed by a destabilized intermediate, with the extent of aggregation being highly specific to both the mAb and the resin type [55].
Table 1: Documented Protein Aggregation in Various Chromatographic Modes
| Chromatography Mode | Protein Studied | Observed Phenomenon | Impact on Recovery/Quality |
|---|---|---|---|
| Cation-Exchange (CEX) | Monoclonal Antibody (mAb) | Multi-peak elution; late-eluting aggregates [55] | Reduced monomer recovery; increased aggregate levels in eluate [55] |
| Reversed-Phase (RPC) | General Proteins | Unfolding and aggregation induced by hydrophobic surfaces [55] | Denaturation and activity loss; multiple peaks of different retention [55] |
| Hydrophobic Interaction (HIC) | General Proteins | Unfolding upon adsorption [55] | Elution of multiple peaks; potential for irreversible aggregation [55] |
| Size-Exclusion (SEC) | General Proteins | Non-ideal interactions with packing material [1] | Altered elution volume; inaccurate molecular weight determination [1] |
Comparative Aggregation Propensity Across Techniques
The propensity to induce aggregation varies significantly between different chromatographic methods, largely dictated by their underlying separation mechanisms and the required operating conditions.
Table 2: Comparison of Aggregation Risk and Key Features Across Major Chromatography Types
| Chromatography Type | Primary Separation Mechanism | Typical Conditions | Relative Aggregation Risk | Key Mitigating Factors |
|---|---|---|---|---|
| Size-Exclusion (SEC) | Molecular size/shape [56] | Aqueous buffer, low viscosity [1] [56] | Low (when no adsorption) [1] | Preserves bioactivity; minimal interaction [56] |
| Ion-Exchange (IEX) | Surface charge [56] | Buffer, variable pH/ionic strength [56] | Low to Moderate [55] | Mild process conditions; sensitive to surface chemistry [55] |
| Hydrophobic Interaction (HIC) | Surface hydrophobicity | High salt concentration | Moderate to High | Salt type and concentration; gradient slope |
| Reversed-Phase (RPLC) | Hydrophobicity [56] | Organic solvent/water gradient [56] | High [55] | Exposure to organic solvents and interfaces [55] |
Mechanistic Insights into Unfolding and Aggregation
Understanding the specific mechanism of protein unfolding on the column is essential for developing effective mitigation strategies. Research suggests that the process often involves multiple steps and can be quantitatively described using kinetic models.
The Three-Point Kinetic Model of Unfolding
A mechanistic model developed to quantify mAb unfolding on a strong CEX medium posits a multi-step process [55]:
- Binding in Native Form: The protein initially adsorbs to the stationary phase in its native, folded conformation.
- Reversible Unfolding: The adsorbed protein undergoes a reversible conformational change, leading to a destabilized intermediate.
- Spreading: The destabilized intermediate may further spread on the adsorbent surface.
- Aggregate Formation: Upon elution, the destabilized intermediate—but not the spread protein—has a high propensity to aggregate [55].
This model successfully reproduces key experimental observations, such as multi-peak elution and reduced recovery, providing a framework for predicting and controlling aggregation during the chromatographic process.
The following diagram details this specific mechanistic pathway, from initial binding to the final formation of aggregates.
Methodologies for Monitoring and Validation
Accurately monitoring and quantifying aggregation is a critical component of any validation protocol. Several established and emerging techniques are employed for this purpose.
Size-Exclusion Chromatography (SEC) as a Gold Standard
Size-exclusion chromatography is the dominant and most reproducible method for the quantitative assessment of protein aggregates, including dimers and higher-order multimers [1]. Its principle is based on the separation of molecules according to their hydrodynamic volume, with larger aggregates eluting first from the column, followed by monomers and smaller fragments [1] [56]. The accuracy of SEC can be confirmed with orthogonal methods like sedimentation velocity analytical ultracentrifugation (SV-AUC) [1]. To ensure validity, SEC methods must be properly calibrated and validated using well-characterized standards to account for potential non-ideal interactions between the protein and the column matrix [1] [7].
Fluorescence-Detection SEC (FSEC)
An advanced variant, Fluorescence-detection Size-Exclusion Chromatography (FSEC), couples the size-based separation of SEC with the sensitivity of fluorescence detection [57]. In this method, the target protein (e.g., a receptor) is fused to a fluorescent protein like GFP. The fluorescence signal directly corresponds to the intact, properly folded protein, allowing for highly sensitive quantification. This approach has been successfully used to detect autoantibodies that reduce the pentameric integrity of receptors, demonstrating its utility in analyzing complex biological interactions [57].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successfully addressing on-column aggregation requires a combination of specialized chromatography materials and analytical tools. The following table lists key solutions used in the featured experiments and this field of research.
Table 3: Research Reagent Solutions for Studying On-Column Aggregation
| Item / Solution | Function / Application | Example Specifications / Notes |
|---|---|---|
| Inert HPLC Columns | Minimizes non-specific adsorption and metal-sensitive interactions for phosphorylated or chelating compounds [58]. | Featuring passivated or metal-free hardware (e.g., Halo Inert, Restek Inert columns) [58]. |
| SEC Columns with Diol Functionalization | High-resolution size-based separation for aggregate analysis with reduced ionic interactions [1]. | BEH particles with diol groups (e.g., SRT SEC-500); usable pH range 2-12, particle sizes down to 1.7 μm for higher efficiency [1] [57]. |
| Cation-Exchange Resins | Studying charge-based separation and pH-gradient induced unfolding/aggregation of mAbs [55]. | Strong cation exchangers; pore size and surface chemistry critically impact aggregate formation [55]. |
| Stable Buffer Systems | Maintaining protein stability and controlling interaction during IEX, SEC, and HIC [1] [55]. | Requires precise pH and ionic strength control; e.g., PBS for SEC mobile phase [1] [56]. |
| Monodisperse MW Standards | Calibrating SEC columns for accurate molecular weight and aggregate quantification [7]. | Certified proteins or polymers; used to generate log M vs. Vr calibration curves [7]. |
| Fluorescent Protein Tags (e.g., GFP, YFP) | Enabling highly sensitive detection in FSEC for tracking receptor integrity and ligand binding [57]. | Fused in-frame with target protein (e.g., α3GFPβ4 nicotinic receptor); stable in lysate for years at -20°C [57]. |
The challenge of on-column protein adsorption and aggregation is a critical consideration in the purification and analysis of biotherapeutics. Evidence shows that even modes considered mild, like IEX, can induce conformational changes and aggregation in specific protein-resin systems [55]. Mitigating these issues requires a strategic approach that includes the selection of appropriate chromatographic modes, the use of inert column hardware to minimize deleterious interactions [58], and careful optimization of elution conditions. Furthermore, employing orthogonal analytical techniques like SEC and FSEC is indispensable for accurately monitoring sample recovery and purity [1] [57]. As the biopharmaceutical landscape evolves to include more complex modalities, continued innovation in column chemistry and a deeper mechanistic understanding of protein-surface interactions will be vital for ensuring high sample recovery and the accurate validation of next-generation analytical sensors.
Using Chemometric Tools and Objective Functions for Systematic Method Optimization
In the fields of analytical chemistry and sensor validation, systematic method optimization is crucial for developing robust, reliable, and efficient analytical procedures. Traditional one-factor-at-a-time (OFAT) approaches, while straightforward, often fail to identify optimal conditions because they cannot account for interactive effects between multiple parameters. This is particularly critical in techniques like size-exclusion chromatography (SEC), used for characterizing natural organic matter (NOM) and validating sensor materials, where mobile phase composition significantly influences separation efficiency through complex physicochemical interactions [59]. Chemometric tools provide a statistical framework for designing experiments, building models, and optimizing multiple variables simultaneously. When combined with objective functions—mathematical expressions that quantify the quality of an analytical outcome—these tools enable researchers to navigate complex experimental spaces efficiently and identify truly optimal conditions, thereby enhancing method performance, reducing development time, and conserving resources [59] [60] [61].
Core Concepts: Objective Functions and Chemometric Tools
The Role of the Objective Function
At the heart of any optimization problem lies the objective function, a mathematical expression that defines the criterion for optimality. In analytical method development, this function, sometimes called a chromatographic response function (CRF), quantifies the quality of an analytical output [59] [60]. The optimizer's goal is to find the set of experimental conditions that either maximize or minimize this function's value.
- Key Components: A typical composite objective function for chromatographic optimization might incorporate several performance criteria. For instance, one developed for SEC of NOM includes the overall resolution between peaks (∑θs,l), the number of distinguishable peaks (N), and the total analysis time (tR,L - t0) [59]. This can be expressed as:
CRF = ∑θs,l + N - (tR,L - t0)/tR,L[59]. - Decision Variables: These are the parameters the experimenter can control, such as mobile phase pH, ionic strength, organic modifier content, and flow rate in SEC [59] [61]. The objective function is a mathematical function of these variables.
- Constraints: These are the limitations imposed on the decision variables, defining the feasible region of experimentation. Examples include the pH range stable for the column packing or the maximum pressure allowed by the chromatographic system [62] [61].
Key Chemometric Tools for Experimentation
Chemometrics provides a suite of statistical techniques tailored for efficient experimental design and data analysis.
- Design of Experiments (DoE): DoE methodologies systematically plan experiments to extract maximum information with minimal runs. A widely used design for optimization is the Central Composite Design (CCD). A CCD efficiently explores the experimental space around a central point by including axial points, allowing for the estimation of quadratic effects and thus, the identification of optimal conditions within the studied domain [59].
- Response Surface Methodology (RSM): Once a DoE is executed, RSM is used to build a mathematical model (often a second-order polynomial) that describes the relationship between the decision variables and the response (the objective function). This model can be visualized as a 3D surface, and the optimum can be readily identified from this surface [59].
- Analysis of Variance (ANOVA): This statistical tool is used to validate the fitted model's significance. It helps determine which factors and interactions have a statistically significant effect on the response, ensuring the model is reliable for prediction and optimization [59].
Comparative Analysis of Optimization Approaches
The choice of optimization strategy profoundly impacts the efficiency and outcome of method development. The table below compares the traditional OFAT approach with the systematic chemometric approach.
Table 1: Comparison of Traditional vs. Chemometric Optimization Approaches
| Feature | Traditional One-Factor-at-a-Time (OFAT) | Systematic Chemometric Approach |
|---|---|---|
| Experimental Design | Unstructured, varying one factor while holding others constant. | Structured using statistical designs (e.g., Full Factorial, CCD). |
| Identification of Factor Interactions | Incapable of detecting interactions between variables. | Explicitly models and identifies significant factor interactions. |
| Number of Experiments Required | Often large and inefficient for complex systems. | Highly efficient, minimizing the number of runs needed. |
| Optimal Condition Prediction | Relies on sequential guessing; may miss the true optimum. | Uses a mathematical model to precisely predict the global optimum. |
| Robustness of Final Method | Method robustness is not systematically evaluated. | Allows for the direct estimation of method robustness. |
| Application Example | Empirical adjustment of SEC mobile phase [59]. | CCD used to optimize pH, [NaCl], and ACN content simultaneously for SEC [59]. |
Application in Size-Exclusion Chromatography and Sensor Validation
A Case Study in SEC Optimization
The principles of chemometric optimization are effectively illustrated by their application to Size-Exclusion Chromatography for characterizing Natural Organic Matter. SEC is prone to non-size exclusion effects (e.g., hydrophobic or ionic interactions), making mobile phase optimization critical [59].
- Experimental Protocol:
- Selection of Factors and Ranges: Key decision variables were identified as mobile phase pH (4.5–7.5), sodium chloride concentration (0–50 mM), and acetonitrile (ACN) content (0–10%). The objective function (CRF) described in section 2.1 was the response [59].
- Experimental Design: A Central Composite Design was employed to structure the experiments, requiring a manageable number of runs to explore the multi-dimensional factor space [59].
- Model Fitting and Optimization: After running the experiments, the resulting CRF data was used to fit a response surface model. This model was then analyzed to find the factor levels that predicted the maximum CRF value, indicating the optimal mobile phase composition for separating the NOM probe compounds [59].
- Outcome: This approach successfully identified a set of robust, optimal conditions (e.g., neutral pH, moderate ionic strength) that minimized undesirable interactions and maximized separation resolution based on molecular size. This methodology can be directly adapted for validating the performance and output of sensors that rely on SEC for analyte separation or characterization.
Essential Research Reagents and Materials
The following table details key reagents and materials required for implementing a chemometrically-optimized SEC method, relevant to both NOM analysis and sensor validation workflows.
Table 2: Essential Research Reagent Solutions for SEC Method Development
| Reagent/Material | Function in SEC Analysis | Example/Chemical Family |
|---|---|---|
| SEC Column | Separates analytes based on their hydrodynamic volume or molecular size. | TSK HW-50S (preparative) [16]; various UHPSEC columns with small particles for high resolution [63]. |
| Probe Compounds/ Standards | Used for system calibration, plate count measurement, and optimization studies. | Polyethylene glycol (PEG), Polystyrene standards, potassium hydrogen phthalate (KHP), monodisperse organic acids [59] [16] [64]. |
| Organic Modifier | Modifies mobile phase to suppress hydrophobic interactions with the stationary phase. | Acetonitrile (ACN), methanol [59]. |
| Buffer Salts | Maintains constant pH to control ionization and suppress ionic interactions. | Phosphate buffer, acetate buffer [59]. |
| Neutral Salt | Adjusts ionic strength to further minimize ionic interactions with the stationary phase. | Sodium chloride (NaCl) [59]. |
| Certified Reference Materials | Validates method trueness, precision, and recovery during method validation. | Suwannee River Fulvic Acid (SR-FA), Pony Lake Fulvic Acid (PL-FA) [59] [16]. |
Experimental Protocols and Data Analysis
Workflow for Systematic Method Development
The following diagram visualizes the end-to-end workflow for applying chemometric tools and objective functions to analytical method optimization.
Systematic Method Optimization Workflow
Protocol for Validating the Optimized SEC Method
After identifying optimal conditions, the method must be thoroughly validated. The following protocol, drawing from thorough validation studies, ensures reliability for routine analysis, such as in drug development or sensor monitoring [16] [65].
- Sample Preservation: Store water samples at 4°C and analyze within two weeks to prevent NOM degradation [16].
- Inorganic Carbon (IC) Removal: Acidify samples to pH 6 using a mild acid (e.g., HCl) and purge with nitrogen gas (N₂) to remove IC without altering the organic composition [16].
- Chromatographic Separation: Use a preparative SEC column (e.g., TSK HW-50S) with a high injection volume (e.g., 1350 μL) to achieve adequate sensitivity without pre-concentration. The mobile phase should be the optimized buffer/salt/organic modifier mixture [16].
- Determination of Key Validation Parameters:
- Linearity: Inject standard solutions (e.g., KHP) across the expected concentration range (e.g., 0.05–10 mg C/L). A coefficient of determination (R²) ≥ 0.998 is typically required [16].
- Limit of Detection (LOD) & Quantification (LOQ): Calculate based on the standard deviation of the response and the slope of the calibration curve. For HPSEC-TOC, LOD can be as low as 19.0 μg C/L [16].
- Precision (Repeatability): Inject the same sample multiple times (n ≥ 6) within a day. Express results as relative standard deviation (RSD). For NOM analysis, RSD can be maintained below 5% [16].
- Trueness (Accuracy): Determine recovery by spiking a real sample with a known quantity of a reference material (e.g., SR-FA) or model compound. Recovery should be close to 100% [16].
- Robustness: Deliberately introduce small variations in method parameters (e.g., flow rate ±0.1 mL/min, temperature ±2°C) to ensure the method output remains unaffected [65].
The integration of chemometric tools and well-defined objective functions represents a superior paradigm for analytical method optimization. Moving beyond traditional OFAT approaches, this systematic framework enables researchers to efficiently develop robust, high-performance methods by quantitatively balancing multiple, often competing, analytical criteria. The application of this approach in SEC, as demonstrated, leads to more reliable characterization of complex materials like NOM, which is fundamental to advancing research in environmental monitoring, drug development, and sensor validation. By adopting these principles, scientists can ensure their analytical methods are not only fit-for-purpose but also operate at their peak predictive capacity and efficiency.
Preventing and Diagnosing Column Degradation for Extended Sensor and System Lifespan
Within the framework of validating self-produced sensors, Size-Exclusion Chromatography (SEC) is an indispensable tool for characterizing the molecular weight and structural integrity of polymeric sensor materials. The accuracy of this validation is critically dependent on the performance of the chromatographic system, particularly the health of the analytical column. Column degradation is an inevitable process wherein the chromatographic performance deteriorates over time due to chemical, thermal, and physical stressors. This degradation directly compromises the quality of separation, leading to inaccurate molecular weight data and potentially invalidating the characterization of synthesized sensors. For researchers and drug development professionals, understanding how to prevent, diagnose, and troubleshoot this degradation is fundamental to ensuring data integrity and extending the operational lifespan of valuable analytical assets.
This guide provides a systematic, evidence-based approach to managing column health. It objectively compares degradation symptoms and prevention strategies across different chromatographic modes, supported by experimental data and detailed protocols. By integrating these practices, scientists can enhance the reliability of their SEC-based validation workflows for novel sensor platforms.
Recognizing Signs of Column Degradation
Early and accurate diagnosis of column degradation is the first step in effective system management. The symptoms can manifest in several ways, often visible in the chromatographic output. The table below summarizes the primary indicators and their common causes, which serve as an initial diagnostic guide.
Table 1: Key Indicators of Column Degradation and Their Common Causes
| Indicator | Description | Common Underlying Cause |
|---|---|---|
| Abnormal Peak Shape [66] [67] | Peak tailing, fronting, or splitting. | Stationary phase degradation, column voiding, or strong unwanted interactions with exposed silanols [66]. |
| Reduced Retention Time [67] | Analytes elute faster than expected. | Loss of stationary phase (e.g., ligand hydrolysis) or the formation of a void at the column inlet [68]. |
| Loss of Resolution [67] | Decreased separation between analyte peaks. | General deterioration of the column's separation efficiency, often from contamination or phase damage. |
| Increased Backpressure | System pressure is significantly higher than the column's baseline. | Blockage of the column frit or tubing by particulate matter or precipitated samples [69]. |
| Unstable Baseline | Noisy or drifting baseline, particularly in GC applications. | Column bleed from degradation of the stationary phase at high temperatures or from oxygen exposure [70]. |
| Unexpected Peaks [66] | Appearance of new peaks not present in initial method runs. | On-column sample degradation catalyzed by active sites on the stationary phase [66]. |
Experimental Observation: On-Column Degradation
The phenomenon of on-column degradation underscores how the column itself can actively alter a sample. In one documented case study, an analyte with an aniline group showed a purity of >95% via NMR but exhibited a chromatogram with a primary peak of only 62% purity and a significant degradant peak [66]. The root cause was traced to interactions with a "lightly loaded" C18 column with high levels of exposed silanol groups on the silica surface [66].
Experimental Protocol for Diagnosis:
- Corroborate with an Orthogonal Technique: When unexpected peaks appear, analyze the sample using a technique like NMR to confirm whether degradation occurred prior to injection [66].
- Systematically Vary Conditions: Shorten analyte exposure time to the column by using a steeper gradient (e.g., starting at a higher organic percentage). A reduction in degradant peaks points to a column-based issue [66].
- Test Alternative Column Chemistry: Repeat the analysis using a high-coverage C18 column from the same manufacturer. The disappearance of degradant peaks on the alternative column confirms the original column's stationary phase was the catalyst [66].
- Modify Mobile Phase Chemistry: Introducing acidic additives (e.g., 0.1% acetic acid) can sometimes stabilize the analyte or pacify active sites on the stationary phase, resolving the degradation [66].
Comparing Degradation Causes and Prevention Across Techniques
Column degradation mechanisms vary depending on the chromatographic technique. The strategies for maximizing column lifespan, therefore, must be tailored to the specific technology, whether it is SEC (a form of LC), GC, or the greener alternative, Supercritical Fluid Chromatography (SFC).
Table 2: Comparative Analysis of Column Degradation: Causes and Prevention Strategies
| Technique | Primary Degradation Causes | Recommended Prevention Strategies | Supporting Data |
|---|---|---|---|
| HPLC/SEC | - Chemical: Mobile phase pH extremes, buffer salts [68].- Physical: Particulate contamination, pressure shocks.- Ligand Loss: Hydrolysis of bonded phases, especially at high T and pH [68]. | - Adhere to manufacturer's pH and temperature guidelines [68].- Use organic buffers (e.g., citrate) over phosphate where possible [68].- Filter samples and use guard columns.- For storage, flush out buffers and store in recommended solvent [68]. | Hybrid silica particles show extended lifetime, maintaining performance for months within pH 1-12, whereas standard silica is limited to pH 2-8 [68]. |
| GC | - Thermal Degradation: Excessive temperatures causing "column bleed" [70].- Chemical: Oxygen exposure, "dirty" samples with non-volatile residues [70]. | - Use high-purity, oxygen-scrubbed carrier gas [70].- Avoid excessive temperatures; use minimal necessary thermal ramp [70].- Implement thorough sample cleanup. Treat the column as a consumable and replace it proactively [70]. | Column bleed is significantly exacerbated by the combination of oxygen and high temperatures. A consistent maintenance schedule (septum changes, scrubber replacement) is critical for prevention [70]. |
| SFC | - Precipitation: Sample or modifier precipitation in the system.- Pressure Fluctuations: Unstable pumping or back-pressure regulation [69]. | - Ensure sample solubility in the CO2/modifier mixture [69].- Optimize the modifier composition and gradient to maintain solubility.- Maintain stable pressure and temperature controls [69]. | SFC is promoted as a "green" technique due to reduced solvent consumption, but its operational stability relies heavily on precise control of the supercritical fluid state [71] [69]. |
A Proactive Toolkit for Column Care and Maintenance
Beyond recognizing symptoms, implementing a rigorous preventative maintenance regimen is the most effective way to extend column lifespan and ensure data quality.
Research Reagent Solutions for Column Maintenance
Table 3: Essential Materials for Column Care and Their Functions
| Item | Function in Column Maintenance |
|---|---|
| In-Line Filter / Guard Column | Protects the analytical column from particulate matter that can clog frits or create voids. |
| High-Purity Buffers & Solvents | Reduces chemical contamination and buildup on the stationary phase. |
| Oxygen/Moisture Trap | (For GC) Removes damaging oxygen and water from carrier gas, suppressing column bleed [70]. |
| Indicating Scrubber | (For GC) Provides a visual signal for when the gas purifier is exhausted and needs replacement [70]. |
| Appropriate Column Storage Solvent | Prevents bacterial growth (for aqueous systems) and ligand hydrolysis during storage. |
Core Preventive Protocols
- Know and Respect Operational Limits: Consult the manufacturer's care and use manual for specific guidelines on pH, temperature, and pressure. Combining pH extremes with elevated temperature is a primary cause of accelerated silica dissolution and ligand hydrolysis [68].
- Ensure Proper Equilibration and Conditioning: Equilibrate the column with 5-10 column volumes of mobile phase. Understand that "conditioning" (e.g., with ion-pairing reagents) is an irreversible process that dedicates the column to a specific assay; avoid unnecessary conditioning for general-purpose columns [68].
- Implement Sample Cleanup: "Dirty" samples leave residues that can bake onto the column, degrading performance. As noted by GC experts, the cost of extensive sample cleanup is often far less than the cost of a prematurely replaced column [70].
- Adopt a Rational Storage Policy: For frequently used reversed-phase columns, storage in the mobile phase is convenient. For long-term storage, flush the column thoroughly to remove all buffer salts and store in a manufacturer-recommended solvent (e.g., high organic content like 80% methanol) [68]. Avoid overly aggressive "washing" protocols, as they can accelerate the removal of hydrolyzed ligand [68].
Diagnostic and Troubleshooting Workflow
When performance issues arise, a systematic approach to troubleshooting is essential. The following diagnostic map guides you through the process of identifying and addressing the root cause of column degradation.
Diagnosis and Action Flowchart
The validation of self-produced sensors via Size-Exclusion Chromatography demands a high degree of confidence in the analytical data produced. As detailed in this guide, proactive column care and systematic diagnostics are not merely maintenance tasks but fundamental scientific practices that underpin data integrity. By recognizing the early signs of degradation, understanding the comparative causes across techniques, and implementing a rigorous preventative toolkit, researchers and drug development professionals can significantly extend the lifespan of their chromatographic systems. This disciplined approach ensures that the characterization of novel sensor materials is both accurate and reliable, thereby supporting the advancement of robust sensor technologies.
Establishing Confidence: Validating Sensor Performance Against Gold Standards
Analytical procedure validation is a critical process in pharmaceutical development and manufacturing, ensuring that test methods are suitable for their intended purpose. The International Council for Harmonisation (ICH) Q2(R2) guideline provides a structured framework for validating analytical procedures used in the release and stability testing of commercial drug substances and products [72]. This guideline outlines key validation characteristics, including accuracy, precision, and linearity, which form the foundation for demonstrating method reliability [72]. For researchers validating self-produced sensors with size-exclusion chromatography (SEC), adhering to this framework provides regulatory alignment while ensuring generated data meets rigorous quality standards.
The following workflow outlines the core stages of the analytical validation process as directed by ICH Q2(R2):
Core Validation Characteristics: Definitions and Experimental Protocols
Linearity
Definition and Regulatory Context: Linearity refers to the ability of an analytical procedure to produce test results that are directly proportional to analyte concentration within a specified range [72]. For SEC methods coupled with novel sensors, demonstrating linearity establishes the quantitative relationship between detector response and analyte amount.
Experimental Protocol:
- Sample Preparation: Prepare a minimum of 5 concentrations spanning the expected working range (e.g., 50-150% of target concentration) [72]. For SEC with evaporative light scattering detection (ELSD), note that the response is typically linear over only one order of magnitude, requiring logarithmic transformation for calibration [73].
- Analysis: Inject each concentration in triplicate using the optimized SEC method.
- Data Analysis: Plot mean response against concentration and perform linear regression analysis.
- Acceptance Criteria: Correlation coefficient (r) typically >0.998 [16], y-intercept not significantly different from zero, and visual inspection of residual plots for random distribution.
Precision
Definition and Regulatory Context: Precision expresses the closeness of agreement between a series of measurements obtained from multiple sampling of the same homogeneous sample under prescribed conditions [72]. It is typically investigated at three levels: repeatability, intermediate precision, and reproducibility.
Experimental Protocol:
- Repeatability: Analyze a minimum of 6 determinations at 100% test concentration on the same day with the same equipment.
- Intermediate Precision: Have a second analyst repeat the study on a different day using different equipment or columns to assess method robustness.
- Data Analysis: Calculate relative standard deviation (RSD%) for each precision level.
- Acceptance Criteria: RSD for repeatability typically ≤1-2% for assay of drug substance, though criteria should be justified based on method purpose [16].
Accuracy
Definition and Regulatory Context: Accuracy expresses the closeness of agreement between the value accepted as a true reference value and the value found [72]. For SEC methods, accuracy validation becomes more complicated when determining molecular weight distribution, requiring specialized approaches [7].
Experimental Protocol:
- Sample Preparation: Prepare samples spiked with known analyte quantities at three concentration levels (e.g., 80%, 100%, 120%) in triplicate.
- Analysis: Analyze samples using the validated SEC method.
- Data Analysis: Calculate percent recovery for each spike level: (Measured Concentration/Known Concentration) × 100.
- Acceptance Criteria: Mean recovery typically 98-102% with RSD ≤2% for drug substance assay [16].
SEC-Specific Validation Considerations for Self-Produced Sensors
Molecular Weight Accuracy Validation
For SEC methods determining molecular weight averages, a specialized accuracy validation approach is required [7]:
- Standard Preparation: Prepare a two-component mixture of monodisperse standards with certified molecular weights that cover the molecular weight range of samples.
- Analysis: Analyze the prepared standard using the SEC method to obtain experimental molecular weight averages ((Mn)exp and (Mw)exp).
- Calculation: Compare experimental results with true values using:
Detector-Specific Considerations for Novel Sensors
Different detection technologies present unique validation challenges, particularly when comparing established detectors to novel sensor technologies:
Table 1: Detector Performance Characteristics in Chromatography
| Detection Type | Dynamic Range | Linearity Range | Key Limitations |
|---|---|---|---|
| Charged Aerosol Detector (CAD) | 4 orders of magnitude [73] | ~2 orders of magnitude [73] | Requires volatile mobile phases |
| Evaporative Light Scattering (ELSD) | 2 orders of magnitude [73] | ~1 order of magnitude [73] | Complex sigmoidal response curves |
| UV-Vis Detection | Varies by analyte | Varies by analyte | Requires chromophores |
| Self-Produced Sensors | To be validated | To be validated | Matrix effects, fouling potential |
Experimental Design and Protocols
Comprehensive Validation Workflow for SEC with Novel Sensors
A systematic approach to validation ensures all critical method attributes are thoroughly assessed:
Sample Preparation and Analysis Protocol
Based on validated SEC methods from literature, the following protocol provides a template for method validation:
- Mobile Phase Preparation: Prepare aqueous SEC mobile phase with 50 mM sodium nitrate and 0.03% sodium azide [36]. Filter through 0.22 μm membrane and degas.
- Standard Solution Preparation:
- Chromatographic Conditions:
- Column: TSKgel G3000SWxl or equivalent SEC column [36]
- Flow rate: 0.5-1.0 mL/min (optimize for separation)
- Injection volume: 10-20 μL (adjust for sensitivity needs)
- Temperature: 25-30°C
- Analysis Sequence: Run blank, system suitability standards, calibration standards, and validation samples in predetermined sequence.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Essential Research Reagents and Materials for SEC Validation
| Item | Function | Example Products/References |
|---|---|---|
| Monodisperse Standards | Calibration for molecular weight determination | Polyethylene glycols, dextrans [75] |
| Certified Reference Materials | Accuracy determination | NIST standards, pharmacopeial reference standards |
| Well-Characterized Polymer Mixtures | Accuracy validation for MW distributions | Two-component mixtures of monodisperse standards [7] |
| High-Purity Mobile Phase Components | Minimize background noise | LC-MS grade solvents, high-purity salts [36] |
| Column Selection Suite | Method optimization | Columns with different pore sizes, materials [36] [16] |
| Stable Isotope-Labelled Analytes | Investigating matrix effects | 13C-labelled compounds for stable isotope-assisted approaches [76] |
Data Interpretation and Acceptance Criteria
Statistical Analysis Methods
Appropriate statistical treatment of validation data ensures objective assessment of method performance:
- Linearity Assessment: Use ordinary least squares regression with evaluation of residual plots. For non-linear detectors like ELSD, consider power function transformations [73].
- Precision Evaluation: Calculate mean, standard deviation, and relative standard deviation (%RSD) for repeated measurements.
- Accuracy Determination: Compute percent recovery and confidence intervals around mean recovery values.
- Comparison Studies: For novel sensors, perform statistical comparison to reference methods using appropriate tests (t-tests, F-tests, ANOVA).
Troubleshooting Common Validation Failures
- Poor Linearity: Check for detector saturation, sample stability, or inappropriate concentration range selection [76].
- Insufficient Precision: Investigate injection technique, column performance, mobile phase preparation consistency, and temperature control.
- Accuracy Deviations: Verify standard preparation, assess matrix effects, and confirm detector linearity in sample concentration range [77].
A rigorously designed pre-study validation protocol for size-exclusion chromatography methods, particularly those employing novel sensors, must address both general ICH Q2(R2) requirements and SEC-specific considerations. By implementing the structured protocols outlined in this guide—with special attention to detector-specific response characteristics, molecular weight accuracy validation, and appropriate statistical analysis—researchers can generate defensible data that demonstrates method suitability for its intended purpose. This validation foundation supports both scientific understanding and regulatory compliance throughout the method lifecycle.
The Analytical Procedure Life Cycle (APLC) provides a holistic framework to ensure analytical procedure fitness for purpose, aligning with Quality by Design (QbD) principles [78]. USP's general chapter <1220> describes a three-stage model, where Ongoing Analytical Procedure Performance Verification (OPPV) constitutes Stage 3, ensuring the procedure remains in a state of control throughout its routine use post-validation [78]. This represents a fundamental shift from treating validation as a "one-off" event to implementing continuous monitoring during everyday operational conditions [78].
For researchers validating self-produced sensors with Size-Exclusion Chromatography (SEC), control charts serve as powerful tools to fulfill OPPV requirements. They provide a statistical framework for monitoring method performance stability, detecting trends, and identifying special cause variation that may indicate method drift or deterioration [79]. This is particularly critical in SEC applications for biologics characterization, where method reliability directly impacts product quality assessment [80].
Control Charts: The Scientist's Toolkit for Method Monitoring
Understanding Control Charts and Process States
Control charts are statistical tools used to monitor, control, and improve processes over time through statistical sampling [79]. They help distinguish between common cause variation (inherent process variation) and special cause variation (non-random, external factors) [79].
Processes can exist in one of four states [79]:
- The Ideal State: In statistical control and produces 100% conformance; predictable and meets customer expectations.
- The Threshold State: In statistical control but produces occasional nonconformance; predictable but doesn't consistently meet customer needs.
- The Brink of Chaos: Not in statistical control but not producing defects; unpredictable but outputs still meet requirements.
- The State of Chaos: Not in statistical control and produces unpredictable nonconformance.
Key Control Chart Elements
All control charts contain three main elements [79]:
- Time Series Graph: The foundational element plotting data points in time order.
- Central Line (X): Represents the process average or location.
- Upper and Lower Control Limits (UCL/LCL): Calculated from process data (typically ±3 standard deviations from the mean), defining the expected range of variation.
Selecting the Appropriate Control Chart
The choice of control chart depends on the data type and context [79]. The following workflow provides a systematic approach for selecting the correct control chart for method performance verification:
Control Charts for SEC Method Performance Monitoring
For SEC method performance verification, specific control charts are particularly relevant:
Individuals and Moving Range (I-MR) Charts: Ideal for monitoring individual SEC runs when the natural subgroup size is unknown, data is scarce, or when monitoring specific performance indicators like retention time, peak area, or molecular weight calculations over time [79]. The I-chart detects trends and shifts in the process average, while the MR chart monitors short-term variability between consecutive observations [79].
Xbar-R Charts: Suitable when rational subgrouping is possible (typically 2-10 observations per subgroup) [79]. For SEC validation, this could involve monitoring the average and range of replicate measurements of quality control samples analyzed within the same batch. The Xbar chart determines the consistency of process averages, while the R chart evaluates the consistency of process variation [79].
Implementing Control Charts for SEC-Sensor Validation: Experimental Protocols
Establishing Control Limits and Baselines
Control limits are calculated from historical process data, not customer specifications [79]. For a new SEC method, initial control limits should be established using data from the method qualification stage (APLC Stage 2) [78].
Protocol: Establishing Initial Control Limits for SEC Performance Indicators
- Data Collection: Collect 20-30 data points of the target performance indicator (e.g., retention time, peak area of a standard, molecular weight calculation) from method qualification experiments [79].
- Control Limit Calculation:
- For I-MR Charts: Calculate the average of the individual values (X̄) and the average moving range (MR̄). Compute control limits as follows [79]:
- I-Chart: UCL/LCL = X̄ ± 2.66 × MR̄
- MR Chart: UCL = 3.27 × MR̄
- For Xbar-R Charts: Calculate the average of subgroup averages (X̄) and the average range (R̄). Compute control limits as follows [79]:
- Xbar Chart: UCL/LCL = X̄ ± A₂ × R̄
- R Chart: UCL = D₄ × R̄, LCL = D₃ × R̄
- For I-MR Charts: Calculate the average of the individual values (X̄) and the average moving range (MR̄). Compute control limits as follows [79]:
- Baseline Assessment: Verify the process is in statistical control during the baseline period before implementing ongoing monitoring.
SEC-Sensor Performance Monitoring Protocol
Experimental Design for Ongoing SEC-Sensor Verification
- Frequency: Incorporate quality control samples at predetermined frequencies based on risk assessment [78]. High-risk methods may require QC samples with each analysis run, while lower-risk methods may require less frequent monitoring [78].
- QC Sample Design: Use stable, well-characterized reference materials that reflect the molecular weight distribution and detector response of actual samples [7].
- Data Collection: Monitor multiple performance indicators simultaneously to comprehensively assess method performance.
Table 1: Key Performance Indicators for SEC-Sensor Method Monitoring
| Performance Indicator | Control Chart Type | Monitoring Purpose | Acceptance Criteria Basis |
|---|---|---|---|
| Retention Time | I-MR Chart | Monitor separation consistency | Method robustness studies |
| Peak Area/Height | I-MR or Xbar-R Chart | Detector response stability | System suitability criteria |
| Theoretical Plates | I-MR Chart | Column performance | Validation data |
| Molecular Weight Averages | I-MR Chart | Separation accuracy | Reference material values |
| Resolution | I-MR Chart | Separation efficiency | ATP/validation criteria |
Data Analysis and Interpretation Protocol
Protocol: Responding to Control Chart Signals
- Out-of-Control Points: Investigate any point outside control limits using root cause analysis [79].
- Trends and Patterns: Apply Western Electric rules or similar pattern recognition guidelines to detect non-random patterns:
- 7+ points in a row on one side of the center line
- 7+ points consistently increasing or decreasing
- Cyclical patterns suggesting environmental or reagent influences
- Reaction Plan: Establish predefined actions for out-of-control situations, which may include [78]:
- Additional analyst training
- SOP review and revision
- Instrument maintenance and qualification
- Method re-optimization or re-validation
Comparative Performance Data: Control Charts vs. Traditional Approaches
Quantitative Comparison of Monitoring Approaches
Table 2: Comparison of Method Performance Monitoring Approaches
| Monitoring Approach | Statistical Basis | Detection Sensitivity | Implementation Complexity | Regulatory Alignment | Best Application Context |
|---|---|---|---|---|---|
| Control Charts (I-MR, Xbar-R) | Statistical process control | High (detects small shifts over time) | Moderate | High (APLC Stage 3, QbD) | Ongoing routine monitoring, trending |
| Periodic Re-validation | Comparison to original validation criteria | Low (point-in-time assessment) | High (resource intensive) | Traditional (ICH Q2) | Major changes, predetermined schedules |
| System Suitability Tests | Pre-set acceptance criteria | Medium (current run only) | Low | Required by regulators | Per-run performance verification |
| Blind Control Monitoring | Comparison to historical data | Medium (requires large dataset) | Medium (requires control management) | Moderate | Long-term precision assessment |
Case Study: SEC Method Performance Assessment
Data from product stability studies can be repurposed to assess SEC method performance. After accounting for the product stability time trend, the remaining variation is attributable to the test method's repeatability and within-lab reproducibility [81].
Table 3: ANOVA Analysis of SEC Method Variation from Stability Data
| Variance Source | Degrees of Freedom | Mean Square | Variance Component | % of Total | Standard Deviation |
|---|---|---|---|---|---|
| Time (Stability) | 1 | 130.32 | - | - | - |
| Lack-of-Fit (Within-lab Reproducibility) | 4 | 17.76 | 4.21 | 45% | 2.05 |
| Replicates (Repeatability) | 12 | 5.134 | 5.14 | 55% | 2.67 |
| Total Method Variation | 17 | - | 9.34 | 100% | 3.06 |
The data in Table 3 demonstrates how variance components analysis can quantify different sources of method variation, with within-lab reproducibility accounting for 45% of total measurement variation in this case [81].
The Analytical Procedure Life Cycle: Integrating Control Charts
The APLC framework provides context for implementing control charts within a comprehensive validation strategy. The following diagram illustrates how control charts integrate across the analytical procedure life cycle:
Risk-Based Approach to Monitoring Frequency and Intensity
The extent of routine monitoring should be defined based on risk assessment, considering procedure complexity, intended purpose, and knowledge about process/procedure variability [78]. A simple risk assessment approach categorizes procedures as [78]:
- Low Risk: Qualitative and semiquantitative tests (e.g., visual tests, simple standard tests, limit tests)
- Medium Risk: Less complex quantitative procedures (e.g., compendial standard tests, water determination, residual solvents)
- High Risk: Complex quantitative procedures (e.g., assay and related substances by LC, bioassays)
For SEC methods used in drug development and quality control, most applications would typically fall into the medium to high-risk categories, justifying comprehensive control chart implementation.
Research Reagent Solutions for SEC Validation
Table 4: Essential Research Reagents for SEC-Sensor Method Validation
| Reagent/Material | Function in Validation | Application Example | Critical Quality Attributes |
|---|---|---|---|
| Monodisperse Standards | Primary calibrants for molecular weight calibration | SEC calibration curve generation | Certified molecular weight, narrow dispersity |
| Polydisperse Reference Materials | Accuracy validation through comparison of experimental vs. actual values [7] | Assessing recovery of molecular weight averages | Well-characterized Mw and Mn values |
| Stable QC Sample | Monitoring system performance over time | Control chart data points | Stability, homogeneity, representative of sample matrix |
| Mobile Phase Components | Maintaining separation consistency | Method robustness assessment | HPLC grade, low UV absorbance, consistent pH |
| Column Storage Solutions | Preserving column performance between runs | Extending column lifetime | Antimicrobial properties, compatible with stationary phase |
Implementing control charts for ongoing method performance verification represents a sophisticated, statistically sound approach that aligns with modern regulatory expectations for analytical procedure life cycle management. For researchers validating self-produced sensors with SEC, this methodology provides objective evidence of method performance throughout its operational lifetime, moving beyond point-in-time validation to continuous assurance of method suitability. Through proper implementation of I-MR, Xbar-R, and other appropriate control charts, scientists can proactively detect method trends, maintain separation consistency, and ensure the reliability of critical molecular weight data used in biopharmaceutical development and quality control.
The validation of novel sensing technologies within rigorous analytical frameworks is a critical step in transitioning them from research prototypes to trusted tools. In the field of size-exclusion chromatography (SEC), a cornerstone technique for biomolecular analysis, the emergence of self-produced sensors promises new levels of customization and cost-effectiveness. This guide provides an objective comparison between these innovative, lab-built sensing platforms and established commercial detectors, contextualized within SEC research for drug development. The performance of a self-validated, self-produced optical sensor for phosphate detection is benchmarked against the capabilities of commercial multi-angle laser light scattering (MALLS) detectors used for absolute molecular weight determination. This analysis aims to equip researchers with the data and methodologies needed to critically evaluate the applicability of self-produced sensors for their specific chromatographic validation needs.
Performance Comparison: Self-Produced vs. Commercial Detectors
The table below summarizes a direct comparison of key performance metrics between a representative self-produced optical sensor and a typical commercial SEC detector system.
Table 1: Quantitative Performance Comparison of Detector Types
| Performance Metric | Self-Produced Fluorescent-Colorimetric Sensor [82] | Commercial MALLS Detector in HP-SEC [83] [84] |
|---|---|---|
| Primary Application | Phosphate detection in aqueous solution | Absolute molecular weight and size of proteins/aggregates |
| Detection Principle | Dual-mode: Fluorescence & Colorimetry | Multi-angle laser light scattering |
| Key Measured Output | Quenching of fluorescence & colorimetric signals | Rayleigh ratio (intensity of scattered light) |
| Measured Analyte | Phosphate (Pi) | Molecular weight (Mw), Radius of gyration (Rg) |
| Selectivity & Validation | Self-validation via dual, independent signal outputs | First-principles measurement (no calibration standards) |
| Limit of Detection | Not explicitly quantified | Not directly applicable (mass-sensitive) |
| Key Advantage | High selectivity and anti-interference; portable platform | Absolute measurement; unaffected by elution volume |
| Documented Limitation | Complex sample matrices may require pretreatment | Higher initial cost; requires complementary concentration detector (UV/RI) |
Detailed Experimental Protocols
Protocol for Self-Produced Sensor for Phosphate Detection
This protocol details the construction and operation of the Ce4+-based dual-mode sensor, which serves as a benchmark for self-produced detectors [82].
Sensor Principle: The sensing mechanism centers on the properties of Ce4+ ions. Dissociated Ce4+ triggers the aggregation-induced emission (AIE) of copper nanoclusters (CuNCs), producing a strong red fluorescence. Concurrently, the Ce4+ acts as an oxidase mimic, catalyzing the oxidation of colorless 3,3′,5,5′-Tetramethylbenzidine (TMB) into a blue-colored product (oxTMB). The introduction of phosphate (Pi) into the system sequesters the Ce4+ ions via a strong P-O-Ce bond, inhibiting both its AIE-triggering and catalytic activities. This results in the quenching of both the fluorescent and colorimetric signals, providing a dual-mode readout [82].
Materials & Synthesis:
- Synthesis of CuNCs: The copper nanoclusters were synthesized using a chemical reduction method. The resulting CuNCs were characterized via transmission electron microscopy (TEM), showing uniformly dispersed particles with a size of 2.7 ± 0.4 nm [82].
- Sensor Assembly: The prepared CuNCs are combined with Ce4+ ions and TMB in a suitable buffer to form the core sensing platform.
Detection Procedure:
- Introduce the aqueous sample containing the target phosphate analyte to the sensor solution.
- Allow the reaction to proceed, during which phosphate binds to Ce4+.
- Measure the fluorescence emission intensity (e.g., at a red wavelength).
- In parallel, measure the UV-Vis absorbance of the solution corresponding to the blue oxTMB.
- The degree of signal quenching in both modes is quantitatively correlated to the phosphate concentration. The two independent signals self-validate the result, ensuring high selectivity against interfering ions [82].
Protocol for Commercial MALLS Detection in SEC
This protocol outlines the standard operation of a commercial Multi-Angle Laser Light Scattering detector coupled with an SEC system for absolute protein characterization [83].
Detection Principle: The technique is based on static light scattering (Rayleigh scattering). As laser light passes through the flowing SEC eluent, molecules in the flow cell scatter the light. The intensity of this scattered light, measured simultaneously at multiple angles, is directly related to the molecular weight and size (radius of gyration) of the solute molecules. The fundamental relationship is described by the Rayleigh equation, allowing for absolute molecular weight determination without reliance on column calibration standards [85] [83].
System Configuration:
- Chromatography System: An HPLC system with an isocratic pump, autosampler, and a size-exclusion column (e.g., Superdex 200).
- Detector Array: The essential setup is a multi-detector array. This typically includes, in sequence:
- A UV absorbance detector and/or Refractive Index (RI) detector to measure the concentration of the eluting protein.
- The MALLS detector itself, which measures the scattered light intensity at multiple angles.
- An optional viscometer to provide intrinsic viscosity data for structural insights [83] [84] [86].
Operation & Data Analysis:
- The protein sample is injected and separated by the SEC column based on hydrodynamic size.
- As each fraction elutes from the column, it passes through the series of detectors.
- The MALLS detector records the light scattering intensity, while the UV/RI detector records the protein concentration at each elution volume increment.
- Software combines the scattering and concentration data using the Rayleigh equation to calculate the absolute molecular weight and size for each data slice across the entire chromatographic peak. This allows for the direct detection of aggregates (higher Mw) and fragments (lower Mw) without the inaccuracies of calibration-based methods [83].
Visualizing the Sensor Validation Workflow in SEC
The following diagram illustrates the logical workflow for validating the performance of a self-produced sensor using the rigorous context of size-exclusion chromatography.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Sensor and SEC Research
| Item | Function/Brief Explanation | Example Context |
|---|---|---|
| Ce4+ ions | Acts as the sensing center in a dual-mode optical sensor; its affinity for phosphate quenches fluorescence and colorimetric signals. [82] | Self-produced phosphate sensor |
| Copper Nanoclusters (CuNCs) | A nanomaterial exhibiting Aggregation-Induced Emission (AIE); serves as the fluorescence source in the sensor. [82] | Self-produced phosphate sensor |
| TMB (3,3',5,5'-Tetramethylbenzidine) | A colorless chromogen that produces a blue color upon oxidation, enabling colorimetric detection. [82] | Self-produced sensor development |
| SEC Columns (e.g., Silica-based) | The stationary phase containing porous beads to separate molecules by hydrodynamic size. [84] | Size-exclusion chromatography |
| Mobile Phase Buffers | Aqueous solvents (e.g., Tris, Phosphate buffers) to maintain protein stability and define elution conditions. [83] | Size-exclusion chromatography |
| Protein Molecular Weight Standards | Well-characterized proteins of known molecular weight for system suitability testing and calibration verification. [83] | SEC-MALLS method validation |
| Static Light Scattering (SLS) Instrument | Measures absolute molecular weight and size (Rg) of polymers/proteins without column calibration. [85] [83] | Commercial SEC detection |
This comparative analysis demonstrates that both self-produced sensors and commercial detectors occupy distinct and valuable niches within the SEC research ecosystem. The self-produced, dual-mode phosphate sensor exemplifies the potential for highly specific, customizable, and cost-effective analytical platforms that can excel in targeted applications, such as monitoring specific metabolites or ions in a sample. Its self-validating nature significantly enhances reliability. In contrast, commercial MALLS detectors offer a robust, off-the-shelf solution for obtaining absolute molecular parameters critical to biopharmaceutical development, directly addressing the limitations of calibration-based SEC.
The choice between these platforms is not a matter of superiority but of strategic alignment with research goals. For researchers focused on a specific analyte where commercial sensors may not exist or are cost-prohibitive, the investment in developing a self-produced sensor is justified. For core facilities and projects requiring unambiguous, absolute characterization of macromolecules, the commercial MALLS detector remains the gold standard. The future of sensing in analytical chemistry likely lies in the convergence of these paths, leveraging the flexibility of self-produced designs with the quantitative rigor of foundational techniques like light scattering, potentially accelerated by data-driven modeling and machine learning for enhanced quality control [87] [88].
In the development and quality control of biopharmaceuticals, the accurate quantification and characterization of protein aggregates are critical. Drug activity, bioavailability, and potential negative side effects, such as adverse immune reactions, can be directly linked to the presence and proportion of aggregates in therapeutic proteins like monoclonal antibodies (mAbs) [89] [90]. For decades, size-exclusion chromatography (SEC) has been the industry standard for this analysis. However, a growing body of literature highlights its limitations, including shear forces that modify samples, adsorption of protein onto the column material leading to low recovery, and a size exclusion limit that restricts the detectable range of aggregates [89] [91]. Consequently, SEC can significantly underestimate aggregate levels, a concern that has prompted regulatory agencies to encourage the use of orthogonal methods to verify SEC results [91].
This guide explores three powerful orthogonal techniques—Analytical Ultracentrifugation (AUC), Asymmetrical Flow Field-Flow Fractionation (AF4), and quantitative PCR (qPCR)—for the comprehensive cross-validation of analytical results. The focus is on their principles, comparative performance, and practical application in validating self-produced sensors and SEC-based research, providing drug development professionals with the data and protocols needed for robust method verification.
Principles of the Orthogonal Techniques
Analytical Ultracentrifugation (AUC): AUC is a first-principle method that separates species based on their sedimentation velocity under a high centrifugal force. It does not rely on a stationary phase or membranes, making it free from interactions that can alter samples. AUC provides a high-resolution size separation and can measure species from monomers up to several hundred million Daltons in molecular weight [91]. However, it has low throughput, requires significant expertise, and does not allow for the collection of fractions for further analysis [89] [91].
Asymmetrical Flow Field-Flow Fractionation (AF4): AF4 separates molecules and particles in an unpacked, thin channel using a perpendicular cross-flow field and the diffusion coefficients of the analytes. Larger species, with smaller diffusion coefficients, are pushed closer to the accumulation wall and elute later than smaller, more diffusive species [92] [93]. This mechanism offers a wide separation range (from ~1 kDa to several MDa), operates without shear forces, and avoids a stationary phase, eliminating associated adsorption issues [89] [94]. It is easily coupled with multiple online detectors and is increasingly recommended as an orthogonal technique by regulatory bodies [89] [90].
Quantitative PCR (qPCR): While not a separation technique, qPCR is a cornerstone molecular technique for the absolute quantification of specific nucleic acid sequences. It operates by monitoring the amplification of a target DNA sequence in real-time using fluorescent probes or dyes. The cycle threshold (Ct) at which the fluorescence signal crosses a defined threshold is used for quantification relative to a standard curve [95]. Its high sensitivity and specificity make it invaluable for applications like detecting microbial contaminants in biological samples [96].
Comparative Performance Data
The following table summarizes a direct comparison of AUC, AF4, and SEC for quantifying aggregates in a monoclonal antibody sample, illustrating a typical scenario where orthogonal methods disagree with SEC.
Table 1: Quantitative Comparison of Aggregate Quantification by AF4, AUC, and SEC
| Analytical Method | Amount of Aggregates Detected (%) | Key Advantages | Key Limitations |
|---|---|---|---|
| Asymmetrical Flow FFF (AF4) | 28.0 | Wide size range (1 kDa - 10 µm); no stationary phase; low shear forces; fraction collection possible [89] [90] | Method development can be complex; potential membrane interactions [92] [91] |
| Analytical Ultracentrifugation (AUC) | 24.0 | High resolution; no matrix interaction; broad size range; operates in formulation buffer [89] [91] | Low throughput; high capital cost; requires skilled operators; no fraction collection [89] [91] |
| Size-Exclusion Chromatography (SEC) | 1.0 | High precision; excellent sensitivity; high throughput; well-established [91] | Stationary phase interactions (adsorption, shear); size exclusion limit; can underestimate aggregates [89] [97] [91] |
This data shows that AF4 and AUC results are in close agreement, while SEC drastically underestimates the aggregate content, likely due to the filtering out or adsorption of aggregates onto the column matrix [89] [90]. A separate study confirmed this trend, finding that both SV-AUC and AF4 detected "markedly higher" aggregate levels than SEC [97].
Table 2: Technique Selection Guide Based on Analytical Needs
| Parameter | AUC | AF4 | qPCR |
|---|---|---|---|
| Primary Measure | Sedimentation coefficient, Molecular weight | Hydrodynamic radius, Molecular weight | Nucleic acid concentration |
| Sample Throughput | Low | Medium | High |
| Size / Dynamic Range | Up to several MDa / > 100 nm [91] | 1 kDa - 10 µm [89] | N/A (target-specific) |
| Key Application in Cross-Validation | Orthogonal check for SEC, high-resolution oligomer separation [91] | Orthogonal check for SEC, complete aggregate profile [89] [91] | Detection of viral/microbial contaminants in cell culture [96] |
Experimental Protocols for Key Techniques
AF4 Method for Monoclonal Antibody Aggregate Analysis
Key Research Reagent Solutions:
- AF4 System: Equipped with an Eclipse AF4 separation channel (Wyatt Technology) and a 10 kDa molecular weight cutoff (MWCO) cellulose membrane [93].
- Mobile Phase: 150 mM ammonium acetate, pH 6.9, filtered (200 nm) and degassed [93].
- Detection System: Online UV detector (280 nm) coupled to a Multi-Angle Laser Light Scattering (MALS) detector for absolute molecular weight determination [89] [92].
Detailed Protocol:
- Channel Preparation: Install a wide spacer (350 µm or 525 µm) and a regenerated cellulose membrane with an appropriate MWCO (e.g., 5-10 kDa) in the AF4 channel [92] [93].
- Focusing/Injection: Inject 20 µL of the mAb sample (approximately 20 µg). Perform a 3-minute focusing step with a focus flow rate of 1.5 mL/min to concentrate the sample into a sharp band at the beginning of the channel [92].
- Elution: Elute the species using a constant or gradient cross-flow (e.g., 2.5-3.5 mL/min) perpendicular to the channel flow. The parabolic flow profile in the channel will separate species based on their diffusion coefficients [92] [93].
- Detection & Analysis: The eluent is directed through a UV detector followed by a MALS detector. The MALS detector allows for the determination of the absolute molar mass and size of the separated monomer and aggregate species without relying on column standards [89] [92].
qPCR Protocol for Specific Nucleic Acid Detection
Key Research Reagent Solutions:
- DNA Extraction Kit: TIANGEN FFPE DNA Kit or equivalent for extracting DNA from complex samples [96].
- qPCR Master Mix: Commercial kit containing DNA polymerase, dNTPs, and buffer.
- Primers and Probes: Specific for target sequences (e.g., IS6110 for M. tuberculosis); a probe for a housekeeping gene (e.g., β-actin) should be included as an internal control [96].
Detailed Protocol:
- DNA Extraction:
- For formalin-fixed paraffin-embedded (FFPE) tissues, deparaffinize five slices (6 µm) by vortexing in 1 mL xylene and centrifuging. Wash the pellet with absolute ethanol and air-dry [96].
- Digest the tissue pellet with proteinase K. Subsequently, purify the DNA using the kit's spin columns and elute in a defined volume [96].
- qPCR Setup:
- Prepare a 25 µL reaction mix per sample containing: DNA template, forward and reverse primers, TaqMan probe, and PCR master mix [96].
- Include no-template controls (blank), negative controls, and positive controls in each run.
- Amplification and Detection:
- Run the plate on a real-time PCR instrument (e.g., SLAN96P) with the following cycling conditions [96]:
- Initial Denaturation: 95°C for 5 minutes.
- 45 Cycles of:
- Denaturation: 95°C for 15 seconds.
- Annealing/Extension: 60°C for 30 seconds (fluorescence acquisition).
- Run the plate on a real-time PCR instrument (e.g., SLAN96P) with the following cycling conditions [96]:
- Data Analysis: Determine the Cycle threshold (Ct) for each sample. A sample with a Ct value greater than 40 is typically considered negative. Quantify the target relative to the standard curve or the internal control [96].
Integrated Workflows and Data Correlation
The true power of orthogonal methods is realized when they are integrated into a cohesive cross-validation strategy. The following diagram illustrates a logical workflow for using AUC, AF4, and qPCR to validate the results from a primary SEC method or a novel, self-produced sensor.
Figure 1: Workflow for Orthogonal Cross-Validation
Interpreting the Correlation Data: As shown in Table 1, a strong correlation between AF4 and AUC results, which simultaneously diverge from SEC data, is a clear indicator that the SEC method may be compromised. This pattern suggests that aggregates are being lost due to shear degradation or adsorption in the SEC column [89] [97]. In this context, AF4 often serves as a "modern alternative" that bridges the gap between the high-resolution but low-throughput AUC and the high-throughput but potentially inaccurate SEC [89] [90]. qPCR functions in a parallel track, providing definitive, sensitive identification of specific nucleic acid targets that are outside the analytical scope of the size-based separation techniques [96].
The correlation of data from orthogonal methods like AUC, AF4, and qPCR is no longer just a best practice but a necessity for robust analytical science in biopharmaceutical development. The experimental evidence consistently demonstrates that reliance on SEC alone can lead to a significant underestimation of critical quality attributes like protein aggregates. AF4 has emerged as a particularly powerful technique, offering a broad analytical range and gentler separation mechanics that provide results comparable to AUC but with greater practicality and throughput. By implementing the integrated workflows and protocols outlined in this guide, researchers and scientists can build a more comprehensive and reliable analytical framework, ensuring the safety and efficacy of therapeutic products through rigorous cross-validation.
The development of bevacizumab biosimilars represents a significant step towards increasing patient access to this critical anti-angiogenic therapy. As a recombinant humanized monoclonal antibody, bevacizumab possesses inherent complexity and heterogeneity, making its analytical characterization particularly challenging [98]. Regulatory agencies worldwide require biosimilar sponsors to demonstrate that their product is "highly similar" to the reference product (Avastin), notwithstanding minor differences in clinically inactive components, and that there are no clinically meaningful differences in terms of safety, purity, and potency [98].
This case study focuses on the application of a validated size-exclusion chromatography (SEC) method for the analysis of bevacizumab in pharmaceutical preparations, framed within the broader context of validating self-produced sensors for biopharmaceutical characterization. The critical role of advanced analytical technologies in comparing biosimilars with corresponding reference products has gained substantial interest, establishing the foundation for biosimilar development requirements [99]. With the U.S. Food and Drug Administration (FDA) increasingly emphasizing the "totality of the evidence" approach, analytical characterization has become the cornerstone of biosimilarity assessments, potentially reducing the need for extensive clinical trials in some cases [100].
SEC Method Validation for Bevacizumab Analysis
Method Principle and Optimization
Size-exclusion chromatography separates proteins based on their hydrodynamic radius, making it particularly valuable for analyzing aggregation in therapeutic proteins [22]. The validated SEC method for bevacizumab analysis employed a Protein KW-804 column (8 × 300 mm) with a mobile phase consisting of phosphate-buffered saline (300 mM NaCl, 25 mM phosphate, pH 7.0) at a flow rate of 1.0 mL/min [22]. The injection volume was set at 25 µL, and detection was performed using a differential refractive index (RI) detector.
Method optimization studies revealed that mobile phase pH significantly impacted peak symmetry. Research demonstrated that tailing factors of the bevacizumab peak improved from 3.1 to 1.3 when the pH of the mobile phase was increased from 6.2 to 6.8 [101]. The selected mobile phase at pH 6.8 yielded maximum signal-to-noise ratio and optimal peak symmetry, critical for accurate quantification.
Pre-study Validation Parameters
A comprehensive pre-study validation was conducted following ICH-Q2(R1) guidelines to demonstrate the method's suitability for its intended purpose [22]. System suitability parameters, including retention time, tailing factors, and theoretical plate number, were measured and found to be within specified limits.
Table 1: SEC Method Validation Parameters for Bevacizumab Analysis
| Validation Parameter | Result | Acceptance Criteria |
|---|---|---|
| Linearity Range | 5-30 µg/mL | R² > 0.99 |
| Precision (RSD) | 0.35% | < 2% |
| Specificity | No interference from excipients | Complete resolution |
| Robustness | Insignificant impact from temperature variations (25-45°C) | System suitability parameters within limits |
| Recovery | Satisfactory across range | 95-105% |
The method demonstrated excellent linearity across the concentration range of 5-30 µg/mL (n=30), with a correlation coefficient satisfying regulatory requirements [22]. The absence of systematic errors was verified by plotting reference versus predicted values for calibration samples. Precision was evaluated by injecting the same sample (30 µg/mL) six times, resulting in an RSD of 0.35%, well within acceptable limits for bioanalytical methods [22].
Coupling with Fluorescence Detection
An advanced SEC method coupled with fluorescence detection has been developed for rapid quantification of bevacizumab in ophthalmic formulations [101]. This approach exploited the intrinsic fluorescence properties of bevacizumab, which shows maximum emission at 340 nm when excited at 280 nm. The method offered several advantages, including reduced analysis time, elimination of organic solvent use, and operation at convenient temperatures that avoid protein destabilization.
The SEC-fluorescence method was validated according to ICH Q2(R1) guidelines and demonstrated satisfactory accuracy, precision, robustness, sensitivity, and specificity, making it suitable for routine analysis of bevacizumab in pharmaceutical dosage forms [101].
Experimental Protocols for Bevacizumab Characterization
Sample Preparation and Chromatographic Conditions
For SEC analysis, bevacizumab samples were prepared to appropriate concentrations using the mobile phase as a diluent [22]. In the case of the fluorescence-coupled method, samples were prepared in phosphate-buffered saline, and the mobile phase consisted of 100 mM sodium phosphate buffer (pH 6.8) containing 300 mM NaCl [101].
The chromatographic system was equilibrated with the mobile phase until a stable baseline was achieved. Separation was performed at room temperature with a flow rate of 1.0 mL/min and detection using either RI or fluorescence detection (excitation 280 nm, emission 340 nm). For the fluorescence-based method, column temperature studies between 25-45°C showed no significant impact on bevacizumab recovery values or tailing factors [101].
Analysis of Forced Degradation Samples
Comparative stability studies of bevacizumab biosimilars involved subjecting formulations to various stress conditions, including thermal stress (elevated temperature of 50 ± 2°C), chemical stress (acidic pH 3.0 ± 0.2, neutral pH 7.0 ± 0.2, and basic pH 10.0 ± 0.2), and mechanical stress (agitation at 200 rpm) [102]. These studies aimed to directly compare the stability of biosimilar formulations with the innovator product.
After exposure to stress conditions, samples were analyzed using SEC to quantify the formation of aggregates and fragments. Additional techniques, including far-UV circular dichroism (CD), were employed to detect alterations in the secondary structure of the native protein [102].
Biosimilarity Assessment Workflow
The analytical workflow for biosimilarity assessment follows a logical progression from comprehensive characterization to targeted quality attribute assessment, as illustrated below:
Comparative Analytical Data: Biosimilars vs. Reference Product
Size Variant and Aggregate Analysis
Multiple studies have employed SEC to analyze the size heterogeneity of bevacizumab biosimilars in comparison to the reference product. A study comparing three bevacizumab biosimilars marketed in India with the innovator product found significant variations in aggregates (p = 0.0306) [103]. The aggregate levels, a critical quality attribute with potential implications for immunogenicity, were quantified using SEC-HPLC.
Another comprehensive study analyzing multiple lots of reference products and their biosimilars included the quantification of high-molecular-weight (HMW) species as a key quality attribute [104]. The researchers measured the extent of distribution in this attribute between products as well as the consistency from lot to lot, providing valuable data on product quality consistency.
Table 2: Comparative Analysis of Bevacizumab Biosimilars and Reference Product
| Product | Acidic Variants (%) | Basic Variants (%) | Aggregates (%) | Biological Activity (%) |
|---|---|---|---|---|
| Reference (Avastin) | Baseline | Baseline | Baseline | 100% |
| Biosimilar A | Significant variation (p<0.0001) | Significant variation (p<0.0001) | Significant variation (p=0.0306) | No significant difference (p=0.6047) |
| Biosimilar B | Significant variation (p<0.0001) | Significant variation (p<0.0001) | Not reported | No significant difference |
| Biosimilar C | Significant variation (p<0.0001) | Significant variation (p<0.0001) | Not reported | No significant difference |
Impact of Analytical Method Variability
The FDA's quality range (QR) method for comparative analytical assessment in biosimilar studies highlights the importance of understanding two key sources of variation: between-lot variation and analytic method uncertainty [22]. The standard deviation of reference product lots depends on both between-lots variation and analytic method uncertainty (within-lots variation).
During the analytical similarity assessment, the method must remain in control and stable with appropriate accuracy and precision. Research indicates that while the SEC method may not always meet the requirements of the Analytical Target Profile (ATP) approach independently of the established uncertainty range, it does satisfy the traditional approach for an uncertainty range of ±2% [22]. This finding underscores the importance of proper method validation and control throughout the biosimilarity assessment process.
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful SEC analysis of bevacizumab requires specific reagents, materials, and instrumentation. The following table details key research solutions essential for conducting these analyses:
Table 3: Essential Research Reagent Solutions for SEC Analysis of Bevacizumab
| Item | Function/Application | Specification/Notes |
|---|---|---|
| SEC Column | Separation based on hydrodynamic radius | Protein KW-804 (8 × 300 mm) or TSKgel G3000SWXL (7.8 × 300 mm, 5 μm) |
| Mobile Phase | Protein separation medium | Phosphate-buffered saline (300 mM NaCl, 25 mM phosphate, pH 7.0 or 6.8) |
| Bevacizumab Reference Standard | System qualification and calibration | Avastin (25 mg/mL) |
| Fluorescence Detector | Sensitive detection of bevacizumab | Excitation: 280 nm, Emission: 340 nm |
| SEC/MALLS | Absolute molecular weight determination | Characterizes aggregation mechanism [22] |
Regulatory Considerations and Future Perspectives
The regulatory landscape for biosimilars is evolving, with agencies like the FDA demonstrating growing confidence in advanced analytical methods. Recent draft guidance from the FDA proposes "major updates to simplify biosimilarity studies and reduce unnecessary clinical testing" [100]. The guidance indicates that comparative efficacy studies (CES) may no longer be routinely required when other evidence, particularly robust analytical data, provides sufficient assurance of biosimilarity.
This regulatory evolution places greater emphasis on the quality and sensitivity of analytical methods such as SEC. The FDA has noted that current analytical technologies can now "structurally characterize highly purified therapeutic proteins and model in vivo functional effects with a high degree of specificity and sensitivity" [100]. Consequently, a comparative analytical assessment (CAA) is generally considered more sensitive than a CES for detecting differences between two products that might preclude a demonstration of biosimilarity.
The relationship between analytical techniques and biosimilar development strategy is multifaceted, encompassing both scientific and regulatory considerations:
The application of a validated SEC method for bevacizumab biosimilar analysis represents a critical component of the comprehensive analytical assessment required for demonstrating biosimilarity. The case study presented herein highlights the importance of rigorous method validation, including demonstration of specificity, linearity, precision, and robustness. As regulatory agencies increasingly recognize the sensitivity of advanced analytical methods in detecting product differences, the role of techniques such as SEC in the biosimilar development pathway continues to expand.
The findings from comparative studies indicate that while bevacizumab biosimilars may show variations in certain quality attributes such as charge variants and aggregate levels compared to the reference product, these differences do not necessarily translate to significant variations in biological activity [103]. This underscores the importance of a holistic approach to biosimilarity assessment that considers the totality of evidence, with SEC playing a pivotal role in characterizing size variants and aggregation profiles.
As the biopharmaceutical industry continues to evolve, the integration of orthogonal analytical methods, including sophisticated SEC approaches coupled with advanced detection systems, will further enhance our ability to comprehensively characterize complex biotherapeutics and facilitate the development of high-quality biosimilars.
Conclusion
The validation of self-produced sensors using Size-Exclusion Chromatography represents a powerful convergence of custom analytical technology and a well-understood separation platform. By adhering to the principles and methods outlined—from foundational knowledge and robust method development to systematic troubleshooting and rigorous validation—researchers can create reliable, cost-effective tools that meet the stringent demands of biopharmaceutical characterization. The future of this synergy is bright, with implications for accelerating biosimilar development, enhancing the analysis of complex therapeutics like AAVs, and bringing sophisticated, multi-attribute monitoring capabilities to a broader range of laboratories. As the field advances, the integration of these sensors with emerging trends such as automation, data-rich hyphenated techniques, and quality-by-design approaches will further solidify their role in the next generation of biomedical analysis.
References
- 1. Size-Exclusion Chromatography for the Analysis of Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3556795/]
- 2. Lecture 04 size exclusion chromatography | PPTX [https://www.slideshare.net/slideshow/lecture-04-size-exclusion-chromatography/237170685]
- 3. Hybrid Particle Columns: The First Twenty Years [https://www.chromatographyonline.com/view/hybrid-particle-columns-first-twenty-years]
- 4. Comparison Techniques for HPLC Column Performance [https://www.chromatographyonline.com/view/comparison-techniques-hplc-column-performance-0]
- 5. Multi-Detection Size Exclusion Chromatography as an ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10254844/]
- 6. Size-exclusion chromatography - Simple English Wikipedia, the free... [https://simple.wikipedia.org/wiki/Size-exclusion_chromatography]
- 7. Accuracy Validation of Size-Exclusion Chromatography [https://www.chromatographyonline.com/view/accuracy-validation-size-exclusion-chromatography]
- 8. Size Exclusion Chromatography (SEC) [https://sse.tulane.edu/polyrmc/instrumentation/sec]
- 9. Size analysis of large DNA molecules by relaxation time ... [https://pubs.rsc.org/en/content/articlehtml/2025/lc/d4lc00998c]
- 10. Thorough Validation of Optimized Size Exclusion ... [https://www.mdpi.com/1420-3049/29/9/2075]
- 11. Size-Exclusion Chromatography of Protein Aggregation in ... [https://www.chromatographyonline.com/view/size-exclusion-chromatography-protein-aggregation-biopharmaceutical-development-and-production]
- 12. Biosensing Strategies to Monitor Contaminants and Additives ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12293825/]
- 13. Trends and new process analytical technologies in ... [https://www.sciencedirect.com/science/article/abs/pii/S037851732500794X]
- 14. The Use of Size Exclusion Chromatography to Monitor ... [https://www.mdpi.com/2073-4352/7/11/331]
- 15. Simple method to analyze the molecular weight of ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400520316452]
- 16. Thorough Validation of Optimized Size Exclusion ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11085112/]
- 17. FAQs: What's the difference between a TOC sensor and ... [https://www.watertechnologies.com/sievers-resource-center/differences-toc-sensor-vs-analyzer]
- 18. Mastering Size Exclusion Chromatography Techniques [https://chromtech.com/blog/mastering-size-exclusion-chromatography-techniques/]
- 19. New Study Compares Wide Pore Size Exclusion ... [https://www.chromatographyonline.com/view/new-study-compares-wide-pore-size-exclusion-chromatography-columns-for-gene-therapy-product-characterization]
- 20. A native SEC-MS workflow and validation for analyzing ... [https://pubmed.ncbi.nlm.nih.gov/38823148/]
- 21. Analytical Similarity Assessment of Biosimilars - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC8865741/]
- 22. Validation of a Size-Exclusion Chromatography Method for ... [https://www.mdpi.com/2297-8739/6/3/43]
- 23. Analytical Assays Determine Biosimilar Product Quality [https://www.biopharminternational.com/view/analytical-assays-determine-biosimilar-product-quality]
- 24. A Strategic Guide to Regulatory Considerations for ... [https://www.drugpatentwatch.com/blog/regulatory-considerations-for-biosimilar-analytical-similarity-assessments/?srsltid=AfmBOor09ijaoKu5epHVGwSmNoDk-0JpzEu0Bve-PcvsXmJ8VwOc0yCj]
- 25. Quantitative description of the quality of size exclusion ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967324004254]
- 26. FAQs on Analysis with Size Exclusion Chromatography [https://www.separations.eu.tosohbioscience.com/technical-support/faqs/analytical-columns/questions-on-sec]
- 27. Size-Exclusion Chromatography of Macromolecules [https://www.mdpi.com/2073-4360/17/5/582]
- 28. Optimizing MS-Compatible Mobile Phases for IEX ... [https://www.chromatographyonline.com/view/optimization-ms-compatible-mobile-phases-iex-separation-monoclonal-antibodies-0]
- 29. Advances in Supercritical Fluid Chromatography for the ... [https://www.chromatographyonline.com/view/advances-supercritical-fluid-chromatography-analysis-chiral-and-achiral-pharmaceuticals]
- 30. Comparison of supercritical fluid chromatographic methods ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967323000833]
- 31. Understanding Sensor Integration in Embedded Systems [https://www.alooba.com/skills/concepts/embedded-systems-225/sensor-integration/]
- 32. How to Quantify the Performance of a Sensor [https://www.airgradient.com/blog/sensor-performance-precision-reproducibility-accuracy/]
- 33. Sensor Integration into Industrial IoT Solutions - Guidelines [https://www.gadgeon.com/blog/sensor-integration-into-industrial-iot-solutions-guidelines/]
- 34. How to Integrate Sensors in IoT - PCBONLINE [https://www.pcbonline.com/blog/sensor-integration.html]
- 35. A Trade-Off Analysis between Sensor Quality and Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9570696/]
- 36. Validation of a high-performance size-exclusion ... [https://www.sciencedirect.com/science/article/pii/S0308814616309943]
- 37. HPLC 2025 Day 1: Automating The Analytical Laboratory [https://www.chromatographyonline.com/view/hplc-2025-day-1-automation]
- 38. Biopharmaceutical Multi-Attribute Method [https://www.thermofisher.com/us/en/home/industrial/pharma-biopharma/pharma-biopharma-learning-center/biopharmaceutical-characterization-information/multi-attribute-method.html]
- 39. Multi-attribute monitoring applications in biopharmaceutical ... [https://www.sciencedirect.com/science/article/pii/S2772391724000537]
- 40. Efficient SEC–UV-RI-MALS Analysis of Protein Aggregates [https://www.chromatographyonline.com/view/efficient-sec-uv-ri-mals-analysis-protein-aggregates-0]
- 41. Method for Improved Fluorescence Corrections for Molar Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9843603/]
- 42. Making chromatography smarter with integrated data ... [https://www.drugdiscoverynews.com/making-chromatography-smarter-with-integrated-data-platforms-16425]
- 43. Pre-study and in-study validation of a size-exclusion ... [https://pubmed.ncbi.nlm.nih.gov/27107247/]
- 44. AAV-SEC Analysis: Top 3 Takeaways From the Latest Article [https://www.thermofisher.com/blog/analyteguru/aav-sec-analysis-top-3-takeaways-from-the-latest-article/]
- 45. Fast and efficient size exclusion chromatography of adeno ... [https://www.sciencedirect.com/science/article/pii/S0021967323008129]
- 46. Comparing analytical approaches for AAV characterization [https://refeyn.com/post/comparing-analytical-approaches-for-aav-characterization]
- 47. Comparison of analytical techniques to quantitate the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8505359/]
- 48. Fast and Ultrasensitive Glycoform Analysis by Supercritical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9685587/]
- 49. A New Approach for Increasing Speed, Loading Capacity ... [https://www.mdpi.com/2227-9717/10/12/2566]
- 50. Optimizing Size-Exclusion Chromatography (SEC) for ... [https://www.chromatographyonline.com/view/optimizing-sec-biologics-analysis-0]
- 51. Self-validating sensor technology and its application in ... [https://www.sciencedirect.com/science/article/abs/pii/S0263224124019109]
- 52. Process monitoring and control using self-validating sensors [https://pureportal.coventry.ac.uk/en/publications/process-monitoring-and-control-using-self-validating-sensors]
- 53. Reentrant liquid condensate phase of proteins is stabilized ... [https://www.nature.com/articles/s41467-021-21181-9]
- 54. Optimizing retention time and peak width reproducibility ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967325002377]
- 55. A case study of the mechanism of unfolding and ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967320309614]
- 56. RPLC vs. HILIC vs. IEX vs. GPC/SEC vs. AC [https://www.metwarebio.com/protein-separation-chromatography-guide-rplc-hilic-iex-gpc-sec-ac/]
- 57. Fluorescence-detection size-exclusion chromatography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12219111/]
- 58. Innovations in Liquid Chromatography: 2025 HPLC ... [https://www.chromatographyonline.com/view/innovations-in-liquid-chromatography-2025-hplc-column-and-accessories-review]
- 59. Optimizing size-exclusion chromatographic conditions ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267011000511]
- 60. Mathematical optimization [https://en.wikipedia.org/wiki/Mathematical_optimization]
- 61. Objective Functions, Decision Variables and Constraints [https://www.baeldung.com/cs/optimization-terminology]
- 62. Objective Function [https://www.geeksforgeeks.org/maths/objective-function/]
- 63. Size-exclusion chromatography-from high-performance to ... [https://pubmed.ncbi.nlm.nih.gov/25116601/]
- 64. Tips & Tricks: Checking the Performance of Your GPC/SEC ... [https://www.chromatographyonline.com/view/tips-tricks-checking-the-performance-of-your-gpc-sec-column]
- 65. An Introduction to Analytical Method Development and ... [https://www.sofpromed.com/an-introduction-to-analytical-method-development-and-validation-in-early-phase-clinical-trials]
- 66. On-Column Sample Degradation [https://www.chromatographyonline.com/view/column-sample-degradation]
- 67. Signs of HPLC Column deterioration and biorelevant media [https://biorelevant.com/learning_center/signs-column-deterioration/]
- 68. Extend LC Column Lifetime: 3 Tips for Chemical Stability [https://www.waters.com/blog/maximizing-lc-column-lifetime/?srsltid=AfmBOoq7N95C8ekDdcvIj4gliKFAHMczlNYhQ6v7N5exyenp-Rfcmvl_]
- 69. Mastering SFC Techniques [https://www.numberanalytics.com/blog/mastering-sfc-techniques]
- 70. GC Column Bleed: Causes and Prevention | Separation Science [https://www.sepscience.com/gc-column-bleed-causes-and-prevention-10132]
- 71. Europe SFC Preview: Sustainability and Supercritical Fluid... 2025 [https://www.chromatographyonline.com/view/sfc-europe-2025-preview-sustainability-and-supercritical-fluid-chromatography]
- 72. ICH Q2(R2) Validation of analytical procedures [https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline]
- 73. CAD vs ELSD: Which HPLC Detector Is Your Better Option? [https://www.thermofisher.com/blog/analyteguru/cad-vs-elsd-which-hplc-detector-is-your-better-option/]
- 74. Comparison of different linear calibration approaches for ... [https://www.sciencedirect.com/science/article/abs/pii/S1570023212006721]
- 75. A SIMPLE SIZE-EXCLUSION CHROMATOGRAPHIC ... [https://chemrxiv.org/engage/chemrxiv/article-details/67b4a4066dde43c9086aa36d]
- 76. Accuracy, linearity, and statistical differences in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11996957/]
- 77. Review and Optimization of Linearity and Precision in ... [https://www.chromatographyonline.com/view/review-and-optimization-linearity-and-precision-quantitative-hplc-elsd-chemometrics]
- 78. Ongoing Analytical Procedure Performance Verification Using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10809227/]
- 79. A Guide to Control Charts [https://www.isixsigma.com/control-charts/a-guide-to-control-charts/]
- 80. Points to Consider in Quality Control Method Validation ... [https://www.bioprocessintl.com/qa-qc/points-to-consider-in-quality-control-method-validation-and-transfer]
- 81. Risk-Based Continued Test-Method Performance ... [https://ispe.org/pharmaceutical-engineering/january-february-2022/risk-based-continued-test-method-performance]
- 82. based self-validated portable platform for highly selective ... [https://www.sciencedirect.com/science/article/abs/pii/S0925400523009607]
- 83. Using Light Scattering Detectors for Size-Exclusion ... [https://www.azom.com/article.aspx?ArticleID=11860]
- 84. High-Performance Size-Exclusion Chromatography [https://www.coriolis-pharma.com/our-services/high-performance-size-exclusion-chromatography/]
- 85. Chromatography Fundamentals, Part IX: Light Scattering ... [https://www.chromatographyonline.com/view/chromatography-fundamentals-part-ix-light-scattering-detectors-for-size-exclusion-chromatography]
- 86. Size Exclusion Chromatography (SEC) [https://www.ornl.gov/content/size-exclusion-chromatography-sec]
- 87. The use of predictive models to develop chromatography- ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9605695/]
- 88. Machine learning anomaly detection of automated HPLC ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d5dd00253b]
- 89. FFF for Quantification of Monoclonal Antibody Aggregates [https://www.news-medical.net/whitepaper/20170510/Advantages-of-Using-Asymmetric-Flow-FFF-for-Quantification-of-Monoclonal-Antibody-Aggregates.aspx]
- 90. Monoclonal Antibody Aggregates [https://www.postnova.com/knowledge/application-notes/application-note/86-id0010_asymmetrical-flow-field-flow-fractionation,-analytical-ultracentrifugation-and-size-exclusion-chromatography-for-monoclonal-antibody-aggregate-quantifi-cation.html]
- 91. Review of Orthogonal Methods to SEC for Quantitation and ... [https://www.biopharminternational.com/view/review-orthogonal-methods-sec-quantitation-and-characterization-protein-aggregates]
- 92. High-Resolution Asymmetrical Flow Field ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6643961/]
- 93. Online Coupling of Asymmetrical Flow Field-Flow ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12232382/]
- 94. Asymmetrical flow field-flow fractionation (AF4) [https://www.coriolis-pharma.com/our-services/asymmetrical-flow-field-flow-fractionation-af4/]
- 95. Frontiers | Droplet digital PCR vs. quantitative real time- PCR for... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1248842/full]
- 96. Clinical performance of quantitative for the molecular... PCR [https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-022-07641-7]
- 97. Quantitation of aggregate levels in a recombinant ... [https://pubmed.ncbi.nlm.nih.gov/17080424/]
- 98. The Art of Biosimilar Reverse Engineering: From Complex ... [https://www.drugpatentwatch.com/blog/the-art-of-biosimilar-reverse-engineering-from-complex-molecules-to-therapeutic-equivalence/?srsltid=AfmBOopW_4L0V3DI2bsT721nOh0ZbhEFGtwAoYMlivBJBcsYLhWtZW4b]
- 99. Analytical tools for characterizing biopharmaceuticals and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3714370/]
- 100. FDA rethinks need for comparative efficacy studies ... [https://www.hoganlovells.com/en/publications/fda-rethinks-need-for-comparative-efficacy-studies-for-certain-biosimilars]
- 101. A validated size exclusion chromatography method ... [https://www.sciencedirect.com/science/article/abs/pii/S073170851930072X]
- 102. Comparative stability study and aggregate analysis of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10558615/]
- 103. Comparative analytical profiling of bevacizumab ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7652956/]
- 104. Characterization of Biosimilar Monoclonal Antibodies ... - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12185665/]