Supercell Lattice Dynamics for Phonon Calculations with Vacancies: A Comprehensive Guide from Foundations to Machine Learning
This article provides a comprehensive exploration of Supercell Lattice Dynamics (SCLD) for investigating phonon properties in materials with vacancy defects.
Supercell Lattice Dynamics for Phonon Calculations with Vacancies: A Comprehensive Guide from Foundations to Machine Learning
Abstract
This article provides a comprehensive exploration of Supercell Lattice Dynamics (SCLD) for investigating phonon properties in materials with vacancy defects. We cover the foundational theory of how vacancies disrupt lattice periodicity and alter phonon character, transitioning from propagating plane waves to localized vibrations and diffusons. The methodological section details practical implementation of SCLD, including supercell construction, force constant extraction, and analysis of phonon broadening. We address common computational challenges and present optimization strategies, notably the integration of machine learning interatomic potentials to accelerate calculations. Finally, the guide covers validation protocols against experimental data and comparative analysis with other computational approaches like the Virtual Crystal Approximation. This resource is tailored for researchers and computational materials scientists working on defect engineering, thermoelectrics, and functional materials design.
Foundations of Disorder: How Vacancies Transform Phonon Physics in Crystalline Materials
{# The Conceptual Framework of SCLD for Defective Systems}
Supercell Lattice Dynamics (SCLD) provides a powerful computational framework for investigating phonon properties in crystalline materials. Traditional lattice dynamics, which relies on the perfect periodicity of an infinite crystal lattice, fails to accurately capture the vibrational behavior of real-world materials containing defects. The SCLD approach overcomes this limitation by using a large, repeated unit cell (the supercell) that explicitly includes defect sites, allowing for the direct calculation of how imperfections like vacancies disrupt harmonic vibrations.
The study of defective systems has moved from a peripheral concern to a central focus in materials science, driven by the understanding that defects can fundamentally alter—and sometimes enhance—a material's properties. Recent research on conjugated coordination polymers (cCPs), for instance, has revealed an advantageous thermoelectric transport regime where electrical conduction is defect-tolerant, but heat propagation is highly defect-sensitive. This combination of high electrical conductivity (up to 2000 S cm⁻¹) with exceptionally low lattice thermal conductivity (down to 0.2 W m⁻¹ K⁻¹) in poorly crystalline structures demonstrates the transformative potential of strategically engineered defects [1].
This Application Note details the conceptual framework and practical protocols for applying SCLD to systems with vacancy defects. It provides researchers with the methodologies to quantitatively link atomic-scale defect structures to macroscopic phonon-influenced properties, enabling the rational design of next-generation materials for applications in thermoelectrics, quantum technologies, and beyond [2] [3].
Theoretical Foundations: From Perfect Crystals to Defective Supercells
The Breakdown of Perfect Crystal Periodicity
In a perfect crystal, the lattice periodicity means the dynamical matrix need only be diagonalized for wavevectors in the first Brillouin zone. The introduction of a vacancy defect breaks this translational symmetry. A vacancy, a zero-mass defect, acts as a strong scatterer for phonons, particularly for high-frequency modes where the wavelength is comparable to the interatomic spacing. This scattering leads to reduced phonon lifetimes and can localize vibrational modes, profoundly impacting thermal conductivity and other vibrational properties [2].
The schematic diagram below illustrates the fundamental conceptual shift from analyzing a perfect crystal lattice to a defective supercell model.
Quantifying Defect-Induced Structural Disorder
The transition from crystalline order to disorder in defective systems is often quantified through parameters like paracrystallinity. Paracrystallinity (g) measures the degree of disorder in a lattice resulting from statistical fluctuations in the position and orientation of unit cells. In Cu₃BHT cCP films, for example, paracrystallinity values exceeding 10% were correlated with a transition to metallic electrical conductivity while maintaining glass-like thermal conductivity—an ideal combination for thermoelectrics [1].
X-ray coherence length, another key metric derived from techniques like GIWAXS, indicates the effective crystallite size within a sample. As defect density increases, this coherence length typically decreases, reflecting the fragmentation of the material into smaller, coherently scattering domains separated by disordered regions or grain boundaries [1].
Computational Methodology and Protocols
SCLD Workflow for Defective Systems
A robust SCLD protocol for systems with vacancies involves multiple stages, from model preparation to the analysis of phonon properties. The workflow must systematically account for defect creation, structural optimization, and the calculation of phonon signatures.
Key SCLD Protocols for Vacancy-Containing Systems
Protocol 1: Defective Supercell Construction and Structural Optimization
- Objective: Create an atomistic model of the material with a controlled concentration of vacancy defects.
- Procedure:
- Start with a fully optimized conventional unit cell of the perfect crystal.
- Construct a supercell of sufficient size (e.g., 10,500 atoms for graphene [2]) to ensure defect separations minimize spurious interactions between periodic images.
- Introduce vacancy defects by removing atoms from specific lattice sites. The defect concentration can be varied systematically (e.g., from 0.1% to 5%).
- Perform full structural relaxation of the defective supercell using Density Functional Theory (DFT) or a suitable classical potential, allowing all atomic positions (and optionally the cell volume) to relax to their new equilibrium positions. This accounts for the local lattice distortion around the vacancy.
- Critical Parameters: Supercell size, vacancy concentration, choice of exchange-correlation functional (for DFT), force convergence criteria.
Protocol 2: Calculation of Phonon Properties Using the Forced Vibrational Method
- Objective: Calculate the phonon density of states (PDOS) and identify localized vibrational modes induced by vacancies.
- Procedure:
- Based on the optimized defective structure, compute the matrix of force constants.
- Apply the Forced Vibrational Method [2], which is efficient for large, complex systems with broken periodicity. This method involves numerically solving the equations of motion for the atoms under periodic boundary conditions.
- Diagonalize the dynamical matrix to obtain the eigenfrequencies and eigenvectors (phonon modes) for the defective system.
- Analyze the PDOS for signature features of vacancies, including peak broadening, softening (shift to lower frequencies), and the appearance of new peaks outside the frequency range of the perfect crystal, which indicate localized modes [2].
- Critical Parameters: Convergence with respect to k-point sampling (if using DFT), numerical accuracy of force constant calculations, free boundary conditions to accurately capture localized modes [2].
Quantitative Data and Property Relationships
Table 1: Quantitative Impact of Defects on Material Properties in Cu₃BHT Films [1]
| Cu/BHT Synthesis Ratio | BHT Vacancy Density (per unit cell) | Paracrystallinity (%) | X-ray Coherence Length (nm) | Electrical Conductivity (S cm⁻¹) |
|---|---|---|---|---|
| 2.0 | 1/3 | 4.8 | 18.5 | 636 ± 245 |
| 3.5 | 1/2 | ~7 | <15 | Up to ~2000 |
| 5.0 | 1/1.8 | ~10 | <10 | Up to ~2000 |
| 6.5 | 1/1.4 | 13 | <8 | N/A |
Table 2: Key Phonon Phenomena in Defective Graphene and Experimental Correlates [2]
| Phonon Phenomenon | Computational Signature | Experimental Correlate | Impact on Properties |
|---|---|---|---|
| Peak Broadening | Broadening of characteristic PDOS peaks (e.g., G peak) | Increased Raman linewidth | Reduced phonon lifetime |
| Peak Softening | Shift of PDOS peaks to lower frequencies | Downshift in Raman peak positions | Altered interatomic force constants |
| Localized Mode Formation | Appearance of new peaks in PDOS outside perfect crystal range | New features in Raman/infrared spectra | Enhanced phonon scattering, reduced thermal conductivity |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Research Reagent Solutions for SCLD and Defect Studies
| Item / Reagent | Function / Role in Research |
|---|---|
| DFT Software (VASP, Quantum ESPRESSO) | Performs first-principles electronic structure calculations to determine total energy, atomic forces, and force constants for the defective supercell. |
| Phonopy Software | A widely used code for calculating phonon properties of periodic solids, capable of handling supercells with defects. |
| Forced Vibrational Method Code | Custom or specialized code for calculating phonon properties in large, fully disordered systems where standard methods struggle [2]. |
| GIWAXS (Grazing-Incidence Wide-Angle X-ray Scattering) | An experimental technique to quantify structural disorder parameters like paracrystallinity and coherence length in thin films [1]. |
| Raman Spectrometer | Measures phonon frequencies and linewidths experimentally; the D band intensity is a direct indicator of defect concentration in sp² carbon materials [2]. |
Application in Advanced Materials Design
The SCLD framework for defective systems is pivotal in explaining and designing materials with tailored thermal and electronic properties. The discovery of defect-tolerant electron transport coupled with defect-sensitive phonon transport in Cu-BHT films exemplifies this [1]. In these materials, charge transport remains efficient even in highly defective structures (paracrystallinity >10%), while phonon transport is severely impeded, leading to lattice thermal conductivities below the amorphous limit. SCLD simulations can unravel the atomic-scale mechanism behind this decoupling, guiding the synthesis of next-generation thermoelectrics.
Furthermore, point defects in wide-bandgap semiconductors like diamond and silicon carbide are the foundation for qubits in quantum technologies [3]. The coherence times of these spin qubits are limited by their interaction with phonons and other spins. SCLD provides critical insights into the electron-phonon coupling and spin-lattice relaxation mechanisms in these systems, enabling the rational selection and design of defect centers with superior quantum properties.
The Supercell Lattice Dynamics framework represents a critical advancement in our ability to model and understand the vibrational properties of real materials. By moving beyond the ideal of the perfect crystal and explicitly incorporating the pervasive influence of defects, SCLD empowers researchers to establish quantitative structure-property relationships in disordered systems. The protocols outlined herein provide a roadmap for systematically investigating how vacancies and other imperfections alter phonon behavior, leading to materials with customized thermal, electronic, and quantum properties. As computational power increases and methods are refined, SCLD will remain an indispensable tool in the quest to harness defects as functional elements in advanced materials design.
In the field of supercell lattice dynamics (SCLD), understanding the fundamental changes in vibrational modes induced by crystal vacancies is crucial for predicting and controlling material properties. This Application Note delineates the theoretical framework and practical methodologies for characterizing how point defects, specifically vacancies, transform vibrational energy carriers from propagons—wave-like propagating phonons—to diffusons—particle-like, diffusive vibrational modes. Within SCLD research, this transition underpins critical phenomena such as anomalous thermal transport, phase stability, and radiation damage response in functional materials.
The presence of vacancies breaks the translational symmetry of the perfect crystal lattice, introducing localized vibrations and scattering channels that fundamentally alter the phonon spectrum. These changes are quantitatively captured through modifications in the interatomic force constants (IFCs), which form the foundation of lattice dynamical calculations [4]. Advanced computational frameworks now enable high-throughput calculation of these anharmonic IFCs, providing a bridge between atomic quantum simulations at 0 K and macroscopic thermal properties at finite temperatures [4].
Theoretical Framework: Vacancy-Induced Perturbations in Lattice Dynamics
Mathematical Foundation of Lattice Vibrations with Defects
The lattice dynamics of a perfect crystal are described by the Taylor expansion of the total energy with respect to atomic displacements, yielding interatomic forces defined by the IFCs [4]:
[{F}{i}^{a}=-\sum _{b,j}{\Phi }{ij}^{ab}{u}{j}^{b}-\frac{1}{2!}\sum _{bc,jk}{\Phi }{ijk}^{abc}{u}{j}^{b}{u}{k}^{c}-\frac{1}{3!}\sum {bcd,jkl}{\Phi }{ijkl}^{abcd}{u}{j}^{b}{u}{k}^{c}{u}_{l}^{d}+\cdots]
Where ({\Phi }{ij}^{ab}), ({\Phi }{ijk}^{abc}), and ({\Phi }{ijkl}^{abcd}) represent the second-, third-, and fourth-order IFCs, respectively; ({u}{j}^{b}) denotes atomic displacements; and ({F}_{i}^{a}) is the resulting force on atom a in direction i.
Vacancies introduce severe local perturbations by removing atoms and their associated bonding interactions, which manifests as:
- Truncated force constant matrices: The removal of an atom eliminates all IFCs associated with that atomic site.
- Local symmetry breaking: The spherical symmetry approximation for vacancy scattering becomes inadequate at high frequencies where the details of the local atomic structure dominate [5].
- Anharmonic enhancement: The strongly asymmetric potential around vacancy sites enhances higher-order anharmonic terms in the force expansion.
Vibrational Mode Transformation Mechanisms
Table 1: Fundamental Mechanisms of Vacancy-Induced Vibrational Mode Transformation
| Mechanism | Effect on Propagons | Effect on Diffusons | Experimental Signature |
|---|---|---|---|
| Localized Mode Creation | Reduces propagating phonon spectral weight | Introduces new resonant frequencies | Additional peaks in VDOS outside host crystal frequency range [6] |
| Elastic Scattering | Shortens phonon mean free path | Increases mode diffusivity | Reduction in thermal conductivity, particularly at low temperatures [5] |
| Phonon Spectral Transfer | Depletes specific frequency ranges | Enhances vibrational density at resonant frequencies | Frequency-dependent suppression and enhancement in VDOS [6] |
| Anharmonic Coupling | Increases phonon-phonon scattering rates | Enhances energy transfer between localized modes | Deviation from classical (T^3) thermal conductivity temperature dependence [7] |
The implantation of a vacancy in a crystal lattice generates three primary types of vibrational perturbations:
- Gap Modes: True localized vibrations with frequencies outside the host crystal's phonon spectrum.
- Resonance Modes: Vibrations with frequencies within the host spectrum but with strongly enhanced vibrational amplitudes near the defect site.
- Hybridized Modes: Mixed character modes resulting from the coupling between propagons and vacancy-induced localized vibrations.
In α-Al₂O₃ with oxygen vacancies, calculations reveal three resonance vibrations in both the acoustic and optical parts of the spectrum with minimal anisotropy in different directions [6]. When the charge state of the vacancy changes, high-frequency optical resonance vibrations emerge, with the frequency of resonance vibrations increasing for F-centers and decreasing for F⁺-centers compared to neutral vacancies [6].
Computational Protocols for SCLD with Vacancies
Workflow for Supercell Lattice Dynamics Calculations
The following diagram illustrates the integrated computational workflow for analyzing vacancy-induced vibrational mode transformations using supercell lattice dynamics:
Supercell Construction and Vacancy Implementation Protocol
Objective: Create computationally efficient supercell models with controlled vacancy concentrations that minimize finite-size effects while maintaining practical computational cost.
Procedure:
- Initial Structure Optimization
- Begin with a fully relaxed primitive cell using DFT with PBEsol functional [4]
- Employ a high plane-wave cutoff energy (≥600 eV) and dense k-point mesh (≥5000 k-points per reciprocal atom)
- Converge total energy to ≤1 meV/atom and forces to ≤1 meV/Å
Supercell Expansion
- Construct supercells with minimum dimension of 20 Å to mitigate vacancy-vacancy interactions [4]
- For high-symmetry materials, use symmetric supercells to maintain point group symmetry
- For low-symmetry systems, employ isotropic supercell expansions
Vacancy Introduction
- Remove single atoms to create monovacancies
- For charged vacancies: Compensate with uniform background charge
- For vacancy clusters: Remove atoms in specific spatial configurations
- Relax atomic positions while keeping supercell vectors fixed
Validation Metrics:
- Convergence of vacancy formation energy with supercell size (target variation <0.01 eV)
- Stability of phonon density of states in the low-frequency region (<2 THz)
- Reproduction of bulk elastic constants within 5% of perfect crystal values
Interatomic Force Constant Extraction Protocol
Objective: Accurately determine harmonic and anharmonic IFCs in vacancy-containing supercells using the finite-displacement method and advanced fitting techniques.
Procedure:
- Atomic Displacements Generation
- Apply displacement amplitude of 0.01-0.03 Å to all atoms within cutoff radius from vacancy
- Use both individual and collective displacements to capture anharmonic terms
- Generate displacement patterns using the non-diagonal supercell method [4]
Force Calculations
- Perform DFT calculations using PBEsol functional with D3 dispersion correction [7]
- Use tighter SCF convergence criteria (10⁻⁸ eV/atom) for displaced structures
- Employ projection operators to separate forces into harmonic and anharmonic components
IFCs Fitting with HiPhive
Quality Control:
- Force fitting accuracy: R² > 0.9 between DFT and fitted forces [4]
- Phonon sum rule enforcement with tolerance <1×10⁻⁴ eV/Ų
- Reproduction of experimental elastic constants within 10%
Table 2: Quantitative Effects of Vacancies on Thermal Properties from SCLD Studies
| Material System | Vacancy Type | Concentration | κ Reduction | Localized Mode Frequency Range | Methodology |
|---|---|---|---|---|---|
| β-SiC [5] | Si monovacancy | 2% | 70% | Not specified | MD-GK (MEAM potential) |
| β-SiC [5] | C monovacancy | 2% | 60% | Not specified | MD-GK (MEAM potential) |
| α-Al₂O₃ [6] | Neutral O vacancy | Single defect | Not measured | Acoustic and optical regions (3 resonances) | Shell model |
| α-Al₂O₃ [6] | F⁺ center (charged) | Single defect | Not measured | High-frequency optical resonances | Shell model |
| BCC Iron [8] | Mono-vacancy | Not specified | Not measured | Not specified | Spin-lattice dynamics |
| Cs₃Bi₂I₆Cl₃ [7] | Not vacancies (lattice distortion) | N/A | Glass-like κ | Not specified | NEP PIMD + WTE |
Analysis Methods for Vibrational Mode Characterization
Phonon Density of States Decomposition Protocol
Objective: Decompose the vibrational density of states into propagons, diffusons, and localized modes to quantify vacancy-induced transformations.
Procedure:
- Spectral Energy Decomposition
- Calculate participation ratio for each phonon mode: (PR(\omega) = \frac{\left(\sumi |ei(\omega)|^2\right)^2}{N\sumi |ei(\omega)|^4})
- Where (e_i(\omega)) is the eigenvector component of atom i for mode frequency ω
- Classify modes: propagons (PR > 0.5), diffusons (0.1 < PR ≤ 0.5), localized modes (PR ≤ 0.1)
Spatial Localization Analysis
- Compute atom-projected phonon density of states (PhDOS)
- Analyze spatial decay of mode amplitudes from vacancy sites
- Identify resonance modes through spectral weight enhancement in local PhDOS
Frequency-Dependent Thermal Transport
- Calculate cumulative thermal conductivity: (\kappa{cum}(\omega) = \int0^\omega \kappa(\omega')d\omega')
- Determine frequency ranges where propagons dominate thermal transport
- Identify suppression frequencies where vacancies most strongly scatter propagons
Thermal Conductivity Calculation Protocol
Objective: Compute the reduction in lattice thermal conductivity due to vacancy-induced phonon scattering using the Wigner transport equation.
Procedure:
- Green-Kubo Formulation
- Calculate heat current autocorrelation function: (\kappa = \frac{V}{3kB T^2} \int0^\infty \langle J(t) \cdot J(0)\rangle dt)
- Where V is volume, T is temperature, and J is heat current vector
- For quantum-corrected results, employ Wigner transport equation [7]
Spectral Thermal Conductivity Decomposition
- Compute mode-dependent thermal conductivity using relaxation time approximation
- Separate contributions from propagons and diffusons
- Quantify scattering rates for different phonon branches
Temperature-Dependent Analysis
- Perform calculations across temperature range (25-300 K) to identify regimes
- Compare perfect crystal and defective supercell results
- Extract vacancy scattering strength from temperature dependence
Research Reagent Solutions: Computational Tools for SCLD
Table 3: Essential Computational Tools for SCLD Studies of Vacancies
| Tool Category | Specific Software/Code | Primary Function | Key Features for Vacancy Studies |
|---|---|---|---|
| DFT Engines | VASP [4] | Electronic structure calculations | PAW pseudopotentials, PBEsol functional [4] |
| Phonon Calculators | Phonopy [4], Phono3py [4] | Harmonic/anharmonic lattice dynamics | Finite-displacement method, mode visualization |
| IFC Fitters | HiPhive [4] | Force constants extraction | Sparse recovery, anharmonic IFCs up to 4th order [4] |
| Thermal Transport | ShengBTE [4], ALAMODE [4] | Thermal conductivity calculations | Wigner transport equation, quantum corrections [7] |
| Machine Learning Potentials | Neuroevolution Potential (NEP) [7] | Accelerated MD simulations | PIMD compatibility, anharmonicity capture [7] |
| Spin-Lattice Dynamics | SLD [8] | Magnetic system simulations | Quantum statistics for phonons and magnons [8] |
Case Study: Oxygen Vacancies in α-Al₂O₃
The following diagram illustrates the vibrational transformations induced by oxygen vacancies in α-Al₂O₃, showcasing the transition from propagons to diffusons:
Application of the protocols outlined in Sections 3-4 to α-Al₂O₃ with oxygen vacancies reveals:
- Neutral Vacancies: Generate three resonance vibrations in both acoustic and optical regions with minimal anisotropy [6]
- Charged Vacancies: F-centers (capturing two electrons) exhibit increased resonance frequencies, while F⁺-centers (capturing one electron) show decreased frequencies compared to neutral vacancies [6]
- Local Phonon Density of States: Shows enhancement at resonance frequencies and suppression in the gap regions between acoustic and optical bands [6]
- Spatial Extent: Localized vibrations demonstrate different decay lengths depending on frequency and charge state
The computational workflow successfully captures these effects through proper supercell construction (≥20 Å), accurate IFC fitting with HiPhive, and detailed phonon DOS analysis. The results demonstrate the transition from propagons to diffusons occurs most strongly in the frequency ranges where resonance modes form, providing a mechanistic understanding of thermal conductivity reduction in defective alumina systems.
This Application Note has established comprehensive protocols for investigating vacancy-induced transformations of vibrational modes using supercell lattice dynamics. The integrated methodology—combining DFT, advanced IFC fitting, and quantum-aware transport calculations—enables quantitative prediction of how vacancies drive the transition from propagons to diffusons across diverse material systems. The case study on α-Al₂O₃ demonstrates the practical application of these protocols, revealing charge-state dependent resonance formation that fundamentally alters vibrational character. These approaches provide researchers with standardized methods for predicting defect-dominated thermal and vibrational properties in functional materials for energy applications, radiation damage assessment, and thermal barrier coatings.
In the field of lattice dynamics, the presence of point defects, such as vacancies, significantly alters the physical properties of materials by disrupting their perfect periodicity. This disruption manifests most clearly in the phonon spectrum, leading to phenomena such as phonon broadening, mode localization, and the emergence of new vibrational features. Supercell lattice dynamics (SCLD) has emerged as a primary computational technique for investigating these effects, allowing researchers to model defective structures and quantify the resulting changes in phonon properties. This Application Note provides a detailed guide to the SCLD methodology, summarizes key quantitative signatures of vacancy-induced disorder, and offers standardized protocols for reproducible research.
Theoretical Background: Phonons in Disordered Systems
In a perfect crystal, the translational symmetry dictates that vibrational normal modes are plane waves characterized by well-defined wavevectors and frequencies. The introduction of vacancies—missing atoms in the lattice—breaks this symmetry. The resulting disorder leads to two primary effects on the phonon spectrum:
- Phonon Broadening: The phonon lifetimes become finite due to scattering from defects, leading to a broadening of the spectral lines. This broadening can be quantified by the full width at half maximum (FWHM) of the phonon peaks in the density of states or dispersion relations [2] [9].
- Localized Vibrational Modes: New vibrational modes can appear outside the frequency range of the perfect crystal. These modes are spatially confined to the region near the defect and are known as localized modes [2].
The character of phonons changes dramatically with the introduction of disorder. Beyond a few percent of impurity concentration, phonons cease to behave as pure plane waves and instead exhibit characteristics similar to the vibrational modes found in amorphous materials. These can be categorized as:
- Propagons: Delocalized, wave-like modes with well-defined group velocities.
- Diffusons: Delocalized modes that do not exhibit a sinusoidal velocity field and conduct heat via diffusion.
- Locons: Modes that are spatially localized due to disorder or defects [9].
Quantitative Data on Vacancy-Induced Phonon Broadening
The following tables summarize key quantitative findings from computational and experimental studies on the effects of vacancy-type defects on phonon properties.
Table 1: Phonon Density of States (PDOS) Changes in Defective Graphene [2]
| Defect Type | Concentration | Observed Effect on PDOS | Quantitative Change |
|---|---|---|---|
| ^13^C Isotope | 1.1% (Natural) | Minimal broadening of characteristic peaks | --- |
| ^13^C Isotope | 50% | Significant broadening and softening of peaks | G peak reduction and broadening |
| Vacancy-type | 0.1% | Emergence of new peaks at ~ 1300 cm⁻¹ | D-band activation |
| Combined ^13^C + Vacancy | Varied | Synergistic broadening and peak softening | Stronger effect than single defect type |
Table 2: Metrics for Characterizing Vibrational Mode Localization [9]
| Metric Name | Formula | Interpretation | Value Range |
|---|---|---|---|
| Participation Ratio (PR) | ( PRn = \frac{\left( \sumi \vec e{i,n}^{\ 2} \right)^2}{N \sumi \vec e_{i,n}^{\ 4}} ) | Measures the fraction of atoms participating in mode (n). Low PR indicates localization. | ( \frac{1}{N} ) (fully localized) to ~1 (fully extended) |
| Eigenvector Periodicity (EP) | Based on velocity field autocorrelation [9] | Classifies modes as propagons, diffusons, or locons based on their spatial character. | Categorical (Propagon, Diffuson, Locon) |
Experimental Protocols for SCLD of Vacancies
Protocol: SCLD Calculation with Band Unfolding for Phonon Broadening
This protocol details the process of using supercell calculations to determine disorder-induced phonon broadening, based on the method demonstrated for binary alloys and defective systems [10] [11].
I. Research Reagent Solutions
Table 3: Essential Computational Tools for SCLD
| Item | Function / Description | Example Software / Potential |
|---|---|---|
| DFT Code | Performs initial structural relaxation and calculates interatomic force constants (IFCs). | VASP, Quantum ESPRESSO, ABINIT |
| Molecular Dynamics Engine | Generates time-dependent atomic configurations at finite temperature. | LAMMPS, GROMACS |
| Lattice Dynamics Code | Diagonalizes the dynamical matrix to obtain phonon frequencies and eigenvectors. | Phonopy, ALM, D3Q |
| Post-Processing Script | Performs the band-unfolding procedure to recover effective dispersion relations. | Custom scripts (e.g., in Python) |
II. Methodology
- Supercell Construction: Generate a sufficiently large supercell of the pristine crystal structure. Introduce a single vacancy by removing one atom. For higher vacancy concentrations, create multiple supercells with vacancies placed in a random, non-interacting configuration.
- Force Constant Calculation: Using a Density Functional Theory (DFT) code, relax the atomic positions of the defective supercell and compute the interatomic force constants (IFCs). Critical Step: Ensure the supercell is large enough so that the IFCs for atoms near the vacancy converge to their bulk values at the supercell boundaries.
- Dynamical Matrix Diagonalization: Construct and diagonalize the dynamical matrix for the defective supercell to obtain its vibrational eigenvalues (frequencies) and eigenvectors (mode shapes).
- Band Unfolding: This key step projects the phonon eigenvectors of the supercell (which has a folded Brillouin zone) back onto the Brillouin zone of the primitive cell. This allows for the calculation of a spectral function, which visualizes the phonon dispersion of the disordered system, showing both the position and broadening of the phonon branches [10].
- Analysis of Broadening: The broadening of the spectral function at a given wavevector is directly related to the phonon lifetime. Analyze the frequency dependence of the broadening and its variation across the Brillouin zone. Calculate the Participation Ratio (PR) for each mode to identify localized vibrations associated with the vacancy [9].
Diagram 1: SCLD Band Unfolding Workflow
Protocol: Forced Vibrational Method for Large-Scale Defective Systems
For very large or complex defective systems where direct diagonalization becomes computationally prohibitive, the Forced Vibrational (FV) method provides an efficient alternative [2].
I. Methodology
- System Setup: Construct a large model system (e.g., N > 10,000 atoms) containing a defined concentration of randomly distributed vacancies.
- Application of Driving Force: Apply an oscillatory external force with a specific frequency ( \omega ) to the system.
- Response Monitoring: Simulate the system's equation of motion and monitor the steady-state amplitude of the atomic vibrations.
- Frequency Sweep: Sweep the driving frequency ( \omega ) across the range of interest.
- Phonon DOS Calculation: The Phonon Density of States (PDOS) is proportional to the square of the vibration amplitude as a function of frequency. The localized modes appear as sharp peaks in the PDOS, while the broadening of bulk modes is directly observable [2].
The Scientist's Toolkit
Table 4: Key Reagents and Materials for Experimental Validation
| Item | Function / Relevance |
|---|---|
| High-Purity Single Crystals | Essential substrate for introducing controlled, quantifiable vacancy concentrations. |
| Ion Implantation System | A primary method for creating vacancy defects by bombarding the crystal with high-energy ions. |
| Annealing Furnace | Used for post-implantation annealing to control vacancy concentration and distribution. |
| Inelastic Neutron Scattering (INS) | A direct experimental technique for measuring the phonon dispersion relation, capable of revealing phonon broadening and soft modes induced by defects [12]. |
| Raman Spectrometer | Probes specific phonon modes; the activation of defect-related modes (e.g., D band in graphene) and linewidth broadening are key signatures of disorder [2]. |
| Momentum-Resolved EELS (q-EELS) | An emerging technique in electron microscopy that allows for mapping of phonon dispersions with high spatial resolution, useful for probing localized phonon behavior [13]. |
Supercell lattice dynamics provides a powerful and versatile framework for quantifying the profound effects of vacancy defects on lattice vibrations. The key spectral signatures—phonon broadening and the emergence of localized modes—can be systematically investigated through the computational protocols outlined herein. As the search results unequivocally demonstrate, even low levels of disorder can radically alter the nature of phonons, moving them from well-defined plane waves to a complex mix of propagons, diffusons, and locons [9]. The integration of robust computational methods like SCLD with advanced experimental probes such as INS and q-EELS is essential for building a predictive understanding of disorder in functional materials, with direct implications for thermoelectrics, battery materials, and radiation damage science.
Translational symmetry, a fundamental principle in crystalline materials, describes the periodic repetition of unit cells in space. The intentional breaking of this symmetry, often through the introduction of defects such as vacancies, creates disruptions in the perfect crystal lattice that profoundly alter a material's vibrational (phonon) and electronic properties. Within the context of supercell lattice dynamics (SCLD), understanding and controlling this symmetry breaking is paramount for predicting and engineering material behavior, particularly in applications ranging from thermoelectrics to quantum information processing. This article explores the theoretical foundations of translational symmetry breaking, its quantitative effects on phonon properties, and provides detailed protocols for its study within SCLD simulations, with a specific focus on vacancy defects.
Fundamental Mechanisms and Material Consequences
Breaking translational symmetry occurs through various mechanisms, each with distinct implications for material properties.
Microscopic Mechanisms of Symmetry Breaking
In condensed matter systems, translational symmetry breaking manifests in several ways. In liquid-crystal coil–rod–coil triblock copolymers, two distinct microscopic mechanisms drive the transition from a nematic to a lamellar phase: one resembles low-dimensional crystallization typical of conventional smectic liquid crystals, while the other is governed by microphase separation due to repulsion between chemically distinct rod and coil segments [14]. In scalar field theory, a soft breaking of translational symmetry can be induced by modifying the standard φ⁴ theory, which also breaks the discrete ℤ₂ symmetry. This mechanism can generate new energy minima and transform kink solutions into asymmetric double-kink configurations, a phenomenon with parallels in dimerized polymer systems like the Su-Schrieffer-Heeger model [15]. In crystalline materials, the most direct form of symmetry breaking comes from point defects. For instance, in URu₂Si₂, a hidden-order phase transition occurs below 17.5 K, accompanied by spontaneous breaking of translational symmetry, as evidenced by the gapping of a heavy quasiparticle pocket observed through angle-resolved photoemission spectroscopy [16].
Consequences for Phonon Properties and Thermal Transport
The introduction of defects that break translational symmetry, particularly vacancies, dramatically impacts lattice dynamics and thermal transport properties. In Zintl-type Sr(Cu,Ag,Zn)Sb thermoelectric compounds, vacancies play a dual role: they not only scatter phonons extrinsically but also profoundly enhance intrinsic lattice anharmonicity. This combined effect results in a giant softening and broadening of the phonon spectrum, causing a remarkable 86% decrease in maximum lattice thermal conductivity compared to the vacancy-free analogue [17]. Similarly, in graphene, the breakdown of translational symmetry through isotope mixing (¹²C to ¹³C) and vacancy-type defects induces phonon scattering and localization. This breakdown activates normally inactive Raman D bands and significantly reduces thermal conductivity by localizing vibrational modes and impeding phonon propagation [2]. The interaction of vacancies with broader lattice dynamics reveals complex behavior; in microtubules, monomer vacancies create seams that act as pre-existing pathways accelerating damage propagation and lattice fracture, with fracture dynamics highly sensitive to the initial defect position relative to these seams [18].
Table 1: Quantitative Impact of Vacancies on Material Properties
| Material System | Type of Symmetry Breaking | Key Consequence | Quantitative Effect |
|---|---|---|---|
| Zintl-type Sr₂ZnSb₂ [17] | Zn-site vacancies (50% concentration) | Lattice thermal conductivity suppression | ~86% decrease (from ~6.82 to ~0.98 W m⁻¹ K⁻¹ at 50 K) |
| Graphene [2] | ¹³C isotope doping & vacancy defects | Phonon scattering & localization | D-band activation in Raman spectra; ~40% reduction in thermal conductivity for 1.1% ¹³C |
| Microtubules [18] | Monomer vacancies creating seams | Accelerated lattice fracture | Fracture time 10-20 minutes; damage span ~1 μm |
| URu₂Si₂ [16] | Hidden-order electronic transition | Gapping of heavy quasiparticle pocket | Formation of "M-shaped" band below 17.5 K transition |
Computational Framework: Supercell Lattice Dynamics with Defects
First-principles calculations, particularly density functional theory (DFT), provide the foundation for modern defect phonon calculations. The supercell approach, where a defect is placed within a periodically repeated cell, is the standard method, albeit computationally demanding for large systems.
High-Throughput Workflow for Lattice Dynamics
An automated high-throughput workflow for lattice dynamics has been developed to systematically calculate phonon properties, including those with significant anharmonicity. This workflow integrates several critical steps and software packages to bridge atomic quantum simulations with macroscopic thermal properties [4]:
- Stringent Structure Optimization: Initial primitive cell optimization and self-consistent field (SCF) force calculations are performed using DFT codes like VASP, employing functionals such as PBEsol that provide accurate lattice parameters for phonon calculations.
- Force Constants Fitting: Harmonic and anharmonic interatomic force constants (IFCs) are fitted using packages like
HiPhive, which efficiently extracts IFCs up to the 4th order from a limited set of strategically perturbed supercells. - Renormalization and Property Calculation: For dynamically unstable compounds, a renormalization step obtains effective phonon spectra at finite temperatures. Properties like vibrational free energy are then calculated using
PhonopyandPhono3py. - Thermal Conductivity Calculation: The lattice thermal conductivity is computed by solving the Boltzmann transport equation using
ShengBTEorPhono3py.
This workflow, implemented in the open-source atomate package, achieves high accuracy (R² > 0.9 for thermal expansion and conductivity across numerous materials) while reducing computational time by 2-3 orders of magnitude compared to conventional finite-displacement methods [4].
Diagram 1: High-throughput workflow for lattice dynamics and thermal property calculation, integrating DFT and machine learning approaches. IFCs: Interatomic Force Constants; κ_lat: Lattice Thermal Conductivity; CTE: Coefficient of Thermal Expansion; F_vib: Vibrational Free Energy.
The "One Defect, One Potential" MLIP Strategy
To overcome the computational bottleneck of large-supercell DFT phonon calculations, a targeted machine learning interatomic potential (MLIP) strategy has been developed. The "one defect, one potential" approach involves training a specific MLIP for a single defect system, yielding phonon accuracy comparable to DFT at a fraction of the cost [19].
Protocol: Training a Defect-Specific MLIP for Phonon Calculations
- Objective: Generate a machine-learned interatomic potential for accurate phonon calculations of a specific point defect in a supercell.
- Software: Neural Equivariant Interatomic Potentials (NequIP) or Allegro packages, which are highly data-efficient due to their E(3)-equivariant architecture [19].
- Procedure:
- Initial Relaxation: Perform a full DFT relaxation of the defect-containing supercell using a converged plane-wave basis set and strict force convergence criteria (e.g., 10 meV/Å).
- Training Set Generation:
- Start from the relaxed structure.
- Generate multiple (e.g., ~40) perturbed supercells by randomly displacing each atom within a small sphere (radius ~0.04 Å) from its equilibrium position.
- For each perturbed structure, calculate the total energy and atomic forces using DFT. This constitutes the training data {Rjα, Fjα, E}.
- Model Training:
- Use 85% of the data for training and 15% for validation.
- Employ a two-body latent multilayer perceptron (MLP) with a cutoff radius of ~6 Å to capture local atomic environments. Training aims to minimize the error between DFT-calculated and MLIP-predicted forces and energies.
- Phonon Calculation:
- Use the trained MLIP with the finite-displacement method (as implemented in
Phonopy) to compute the force constants matrix for the supercell. - The MLIP predicts forces for each displaced structure in seconds, dramatically accelerating the process compared to DFT.
- Use the trained MLIP with the finite-displacement method (as implemented in
This method reduces computational expenses by over an order of magnitude while maintaining high accuracy for derived properties like Huang–Rhys factors, photoluminescence spectra, and nonradiative capture rates [19].
Diagram 2: The "one defect, one potential" strategy workflow for training a machine learning interatomic potential (MLIP) tailored for accurate defect phonon calculations.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 2: Key Computational Tools and Resources for Defect Phonon Studies
| Tool/Resource | Type | Primary Function in SCLD | Application Example |
|---|---|---|---|
| VASP [19] [4] | DFT Software | Provides reference total energies and atomic forces for training data and direct phonon calculations. | Structure relaxation and force calculations for defect supercells. |
| Phonopy [19] [4] | Phonon Analysis Code | Implements the finite-displacement method and processes force constants to obtain phonon frequencies and densities of states. | Post-processing forces from DFT or MLIP to get defect phonon spectra. |
| HiPhive [4] | Force Constant Fitter | Extracts harmonic and anharmonic IFCs from a limited set of supercell displacements using advanced regression techniques. | Building effective force constants for thermal property calculation in high-throughput workflow. |
| Allegro/NequIP [19] | MLIP Code | Constructs equivariant neural network potentials that are highly data-efficient for learning complex potential energy surfaces. | Training a defect-specific potential for rapid force predictions in large supercells. |
| ShengBTE/Phono3py [4] | Thermal Property Calculator | Solves the Boltzmann transport equation for phonons to compute lattice thermal conductivity, including three-phonon scattering. | Calculating κ_lat reduction due to vacancy scattering and anharmonicity. |
The breaking of translational symmetry through defects like vacancies is a powerful mechanism for tailoring material properties, particularly phonon behavior. The theoretical underpinnings of this phenomenon span from modified field theories to practical defect engineering in crystals. The computational framework of supercell lattice dynamics, now supercharged by high-throughput workflows and targeted machine learning potentials, provides the essential tools to probe these effects with unprecedented accuracy and efficiency. The "one defect, one potential" strategy represents a paradigm shift, enabling the study of defect phonons in large supercells that were previously computationally prohibitive. These advanced protocols empower researchers to systematically design materials with customized vibrational and thermal properties for applications in thermoelectrics, photonics, and quantum technologies.
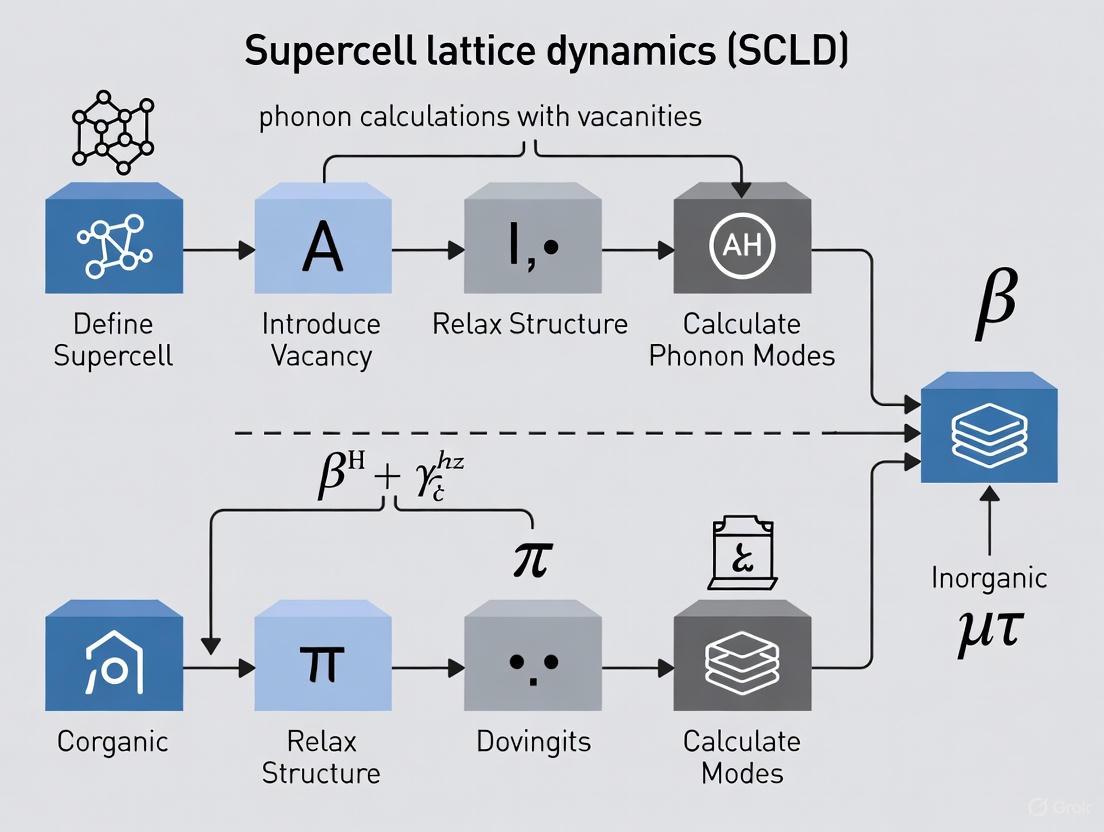
Practical Implementation: A Step-by-Step Guide to SCLD Calculations with Vacancies
The supercell approximation is a foundational technique in computational materials science for simulating non-ideal systems, including those with point defects like vacancies, within otherwise periodic crystalline materials. This approach enables the application of standard periodic boundary condition calculations to disordered systems by creating a sufficiently large periodic cell that replicates the local structural properties of the real, disordered material at the atomic level. For research in supercell lattice dynamics (SCLD), particularly for phonon calculations in systems with vacancies, two strategic challenges are paramount: determining the minimal supercell size that ensures converged lattice dynamical properties and implementing a methodical approach for sampling the numerous possible vacancy configurations. This application note details rigorous protocols for addressing these challenges, enabling reliable and computationally efficient simulations of defective materials.
Supercell Size Convergence for Phonon Calculations
The Core Principle: Isolating Defect-Defect Interactions
In a perfect crystal, the primitive cell is sufficient for calculating phonon properties. Introducing a vacancy, however, breaks the crystal's perfect periodicity. The supercell method restores periodicity by repeating a unit cell containing the defect. The central goal in size convergence is to make the supercell large enough that the interactions between a vacancy and its periodic images become negligible. This ensures the calculated properties, such as the force constants and vibrational frequencies, approximate those of an isolated defect.
Quantitative Criteria for Size Selection
The table below summarizes the key physical and computational criteria for determining a sufficiently converged supercell size.
Table 1: Criteria for Supercell Size Convergence in Phonon Calculations
| Criterion | Description | Quantitative Target |
|---|---|---|
| Force Constant Decay | The force constant matrix elements must decay to approximately zero within the supercell dimensions. | A supercell size where the magnitude of force constants falls below a negligible threshold (e.g., < 1% of its maximum value) at the supercell boundary [20]. |
| Physical Cutoff Radius | The minimum supercell dimension should be larger than twice the cutoff radius of the force constant decay. | Supercell side length > 15 Å is often a practical starting point, as force constants in many materials decay to near-zero within ~7.5 Å [20]. |
| Vacancy Concentration | The effective concentration of vacancies in the supercell should be low enough to mimic an isolated defect. | Target concentrations of < 2% are often necessary, requiring supercells of at least 50 atoms for a single vacancy [21]. |
| q-Point Grid Equivalence | A supercell of size N₁×N₂×N₃ is equivalent to sampling the Brillouin zone with a q-grid of the same dimensions. | A converged q-grid (e.g., 4×4×4 or finer) is typically required, dictating the minimum supercell size [20]. |
Recommended Protocol: Convergence Testing
A systematic convergence test is the only reliable method to determine the appropriate supercell size for a specific system. The workflow below outlines this essential procedure.
Protocol Steps:
- Initialization: Begin with a supercell that meets the minimum physical size (e.g., all dimensions > 15 Å) and corresponds to a reasonably coarse q-point grid (e.g., 3×3×3).
- Calculation: For the chosen supercell, perform a full geometry relaxation to find the ground-state structure in the presence of the vacancy, followed by a phonon calculation (using methods like density functional perturbation theory or finite displacement as in Phonopy).
- Property Extraction: Calculate key properties sensitive to supercell size, most importantly the vacancy formation energy and the vibrational frequencies of modes localized near the vacancy.
- Iteration: Increase the supercell size significantly (e.g., to 4×4×4). For non-cubic systems, consider using non-diagonal supercells [20], which can achieve the same q-point sampling with fewer atoms, drastically reducing computational cost.
- Convergence Check: Compare the target properties from the new, larger supercell with the previous one. Convergence is typically achieved when the change in vacancy formation energy is less than 0.01 eV and the phonon frequencies of interest shift by less than 0.1 THz.
Vacancy Configuration Sampling
The Combinatorial Challenge
When multiple vacancies are present in a supercell—either to model higher defect concentrations or to capture vacancy-vacancy interactions—a single configuration is insufficient. The arrangement of these vacancies within the supercell can significantly influence the calculated lattice dynamical properties. The goal of sampling is to explore a representative set of these unique, symmetry-inequivalent configurations.
Mathematical Foundation and Statistics
For a supercell with ( K ) equivalent sites and ( v ) vacancies to be placed, the total number of possible configurations is given by the binomial coefficient:
[
P_{\text{total}} = \binom{K}{v} = \frac{K!}{v!(K-v)!}
]
However, many of these configurations are symmetrically equivalent. The number of unique configurations is reduced by the symmetry operations of the supercell's space group. The supercell program [21] and similar algorithms [22] are designed to generate only these symmetry-inequivalent structures.
Table 2: Configuration Statistics for a 3×3×3 FCC Supercell (108 sites)
| Number of Vacancies (v) | Total Configurations | Approximate Unique Configurations |
|---|---|---|
| 1 | 108 | ~10-30 |
| 2 | 5,778 | ~100-500 |
| 3 | 205,284 | ~1,000-5,000 |
Recommended Protocol: Systematic Configuration Sampling
The following workflow ensures a comprehensive and manageable sampling of vacancy configurations.
Protocol Steps:
- System Definition: Specify the supercell, the number of vacancies, and their type.
- Configuration Generation: Use a tool like the
supercellprogram [21] to generate all symmetry-inequivalent vacancy configurations. This software applies combinatorial algorithms and space group symmetry operations to list unique structures. - Configuration Filtering: Apply physical filters to reduce the set to the most physically plausible configurations.
- Minimum Vacancy-Vacancy Distance: Impose a cutoff to avoid unphysically high repulsive interactions.
- Coulomb Energy (for charged systems): Calculate the Ewald energy and prioritize configurations with lower electrostatic energy [21].
- Representative Subset Selection: For complex systems with hundreds of unique configurations, select a representative subset (10-20) that captures the diversity of vacancy arrangements, focusing on those with the lowest energies.
- Calculation and Analysis: Perform phonon calculations on the subset of configurations. Analyze the results by calculating the average and standard deviation of key properties (e.g., specific phonon frequencies, thermodynamic properties). The standard deviation quantifies the property's sensitivity to configurational disorder.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Software and Computational Tools for SCLD with Vacancies
| Tool / Solution | Function | Relevance to SCLD with Vacancies |
|---|---|---|
supercell program |
An all-in-one software for generating supercells with point defects and sampling atomic configurations [21]. | Core tool for implementing the vacancy configuration sampling protocol. It handles symmetry analysis, combinatorics, and charge balancing. |
| Phonopy | A widely used open-source package for calculating phonon spectra and properties [20]. | Primary engine for performing the finite-displacement phonon calculations on the generated supercells containing vacancies. |
| Non-Diagonal Supercells | A mathematical approach to construct compact supercells for a given q-point grid [20]. | Dramatically reduces computational cost in convergence testing by enabling finer q-point sampling with fewer atoms. |
| DFT Codes (VASP, Quantum ESPRESSO, ABINIT) | First-principles electronic structure codes using Density Functional Theory. | Used for the underlying force and energy calculations required for geometry relaxation and phonon analysis. |
| Special Quasi-random Structures (SQS) | A method to generate a small number of supercells that best represent a random alloy [21]. | An alternative, less exhaustive sampling strategy for modeling very high, random vacancy concentrations. |
The finite-displacement method, also known as the supercell method, small-displacement method, or frozen-phonon approach, represents a cornerstone technique in computational materials science for calculating the vibrational properties of crystalline solids [23]. This approach is fundamentally based on the theory of lattice dynamics, which studies the collective atomic vibrations in a crystal, quantized as phonons. Within the framework of density-functional-theory (DFT) based first-principles calculations, the finite-displacement method operates by introducing small, controlled displacements of atoms from their equilibrium positions within a supercell—a larger periodic cell constructed by repeating the primitive unit cell [23]. The resulting forces on all atoms are computed quantum-mechanically, enabling the extraction of interatomic force constants (IFCs) that parameterize the potential energy surface in the harmonic approximation.
For imperfect lattices containing point defects such as vacancies, the method's implementation requires particular care. The presence of vacancies breaks the perfect periodicity of the crystal, necessitating the use of sufficiently large supercells to isolate the defect and prevent unphysical interactions between its periodic images [21]. The core objective is to determine the real-space interatomic force constant matrix ( \Phi_{\alpha\beta}^{jk}(P, Q) ), which describes the interaction between the ( j )-th atom in primitive unit cell ( P ) and the ( k )-th atom in primitive cell ( Q ) along Cartesian directions ( \alpha ) and ( \beta ) under zero macroscopic electric field [23]. These force constants are foundational for constructing the dynamical matrix, whose diagonalization yields phonon frequencies and eigenvectors throughout the Brillouin zone, ultimately enabling the prediction of thermal properties such as free energy, heat capacity, and lattice thermal conductivity.
Theoretical Foundation and Computational Framework
Fundamental Equations of Lattice Dynamics
Within the harmonic approximation, the potential energy ( E(U) ) of a displaced crystal is expanded to second order in the atomic displacements ( u_\alpha(t, j; P) ) from their equilibrium positions [23]:
[ E(U) = \frac{1}{2} \sum{P,Q}^{N} \sum{j,k}^{NP} \sum{\alpha,\beta}^{3} \Phi{\alpha\beta}^{jk}(P, Q) u\alpha(t, j; P) u_\beta(t, k; Q) ]
Here, ( N ) is the number of primitive unit cells in the supercell, ( NP ) is the number of atoms in the primitive cell, and ( \Phi{\alpha\beta}^{jk}(P, Q) ) is the real-space interatomic force constant matrix. The force on atom ( (j, P) ) in direction ( \alpha ) is obtained as the negative gradient of the potential energy:
[ F{\alpha}(j, P) = - \frac{\partial E(U)}{\partial u\alpha(j, P)} = - \sum{Q,k,\beta} \Phi{\alpha\beta}^{jk}(P, Q) u_\beta(k, Q) ]
In the finite-displacement method, these forces are computed directly from first-principles calculations for a set of small, finite displacements ( {\delta u} ), and the force constants are extracted by inverting the above relationship. For a polar solid, an additional complication arises: certain optical vibration modes create dipole-dipole interactions and homogeneous electric fields. The force equation must then include a nonanalytic correction [23]:
[ mj \frac{\partial^2 u\alpha(t, j; P)}{\partial t^2} = - \frac{\partial E(U)}{\partial u\alpha(t, j; P)} + e Z\alpha(j) \cdot E ]
where ( mj ) is the atomic mass, ( e ) is the electron charge, ( Z\alpha(j) ) is the Born effective charge tensor, and ( E ) is the vibration-induced macroscopic electric field. This term is responsible for the LO-TO splitting—the removal of degeneracy between longitudinal optical (LO) and transverse optical (TO) phonons at the Brillouin zone center [23].
The Supercell Approximation for Disordered Systems
The supercell approximation is the primary strategy for applying periodic boundary conditions to non-periodic systems such as imperfect lattices with vacancies [21]. In this approach, a large supercell is constructed that reflects the local structural properties of the disordered system as closely as possible within its boundaries. For vacancy defects, this involves creating a supercell from the perfect crystal's unit cell and removing one or more atoms to create vacant sites.
The key challenge lies in generating physically meaningful configurations of these defects. When the concentration of vacancies is high, their relative positions and interactions become critical. An exhaustive combinatorial approach can be employed where all unique symmetry-independent configurations of vacancies within the supercell are generated and processed [21]. The number of unique configurations grows rapidly with supercell size and vacancy concentration, necessitating efficient symmetry analysis algorithms to avoid redundant calculations.
Table 1: Key Parameters in Supercell Construction for Vacancy Studies
| Parameter | Description | Considerations for Vacancies |
|---|---|---|
| Supercell Size | Number of primitive unit cells used to create the supercell | Must be large enough to isolate vacancy and prevent spurious interactions between periodic images |
| Defect Concentration | Ratio of vacant sites to total sites in the supercell | Lower concentrations require larger supercells for accurate representation |
| Symmetry | Space group symmetry of the supercell with vacancies | Reduced symmetry compared to perfect crystal; symmetry operations used to identify equivalent configurations |
| Charge State | Net charge of the supercell due to vacancy creation | May require charge compensation through background charge or explicit counter-defects |
Computational Protocols and Methodologies
Workflow for Force Constant Extraction in Imperfect Lattices
The following diagram illustrates the comprehensive workflow for implementing the finite-displacement method in imperfect lattices containing vacancies:
Workflow for Force Constant Extraction
Step-by-Step Implementation Protocol
Begin with the optimized crystal structure of the perfect material. Generate a supercell of sufficient size to minimize vacancy-vacancy interactions through periodic boundary conditions. The required size depends on the vacancy concentration and the range of interatomic interactions—typically 2×2×2 to 4×4×4 multiples of the conventional unit cell. Using the supercell program or similar tools, systematically introduce vacancies at the desired crystallographic sites [21]. For a comprehensive study, generate all symmetry-inequivalent configurations using combinatorial algorithms:
[ P(k1, k2, \ldots, kN) = \frac{\left(\sum{i=1}^N ki\right)!}{\prod{i=1}^N k_i!} ]
where ( k_i ) represents the number of atoms of type ( i ) on the disordered site, with vacancies treated as a special "null" atom type [21].
Step 2: Structural Relaxation and Electronic Structure Calculation
For each unique vacancy configuration, perform a full structural relaxation using DFT to allow atomic positions and cell parameters to adjust to the defect presence. Employ a plane-wave basis set with appropriate pseudopotentials, ensuring a high enough energy cutoff and k-point sampling for convergence. Calculate the electronic structure to determine the defect's charge state and any possible mid-gap states introduced by the vacancy.
Step 3: Atomic Displacements and Force Calculations
Implement a displacement pattern where each symmetrically inequivalent atom in the supercell is displaced by a small amount (typically 0.01-0.03 Å) in positive and negative directions along each Cartesian axis. For each displacement, perform a single-point DFT calculation to obtain the resulting forces on all atoms in the supercell. The force constant matrix elements can be approximated through the central finite-difference formula:
[ \Phi{\alpha\beta}(i,j) \approx - \frac{F\alpha(i, \delta u\beta(j)) - F\alpha(i, -\delta u\beta(j))}{2\delta u\beta(j)} ]
where ( F\alpha(i, \delta u\beta(j)) ) is the force on atom ( i ) in direction ( \alpha ) when atom ( j ) is displaced in direction ( \beta ).
Step 4: Force Constant Extraction and Symmetrization
Collect the force response matrices for all displacements and construct the full force constant matrix. Apply the appropriate symmetry operations to ensure the force constants obey the physical constraints of translational and rotational invariance:
[ \sumj \Phi{\alpha\beta}(i,j) = 0 \quad \text{(translational invariance)} ] [ \sumj \Phi{\alpha\beta}(i,j) r\gamma(j) - \Phi{\alpha\gamma}(i,j) r_\beta(j) = 0 \quad \text{(rotational invariance)} ]
where ( r_\gamma(j) ) represents the γ-coordinate of atom ( j ).
Step 5: Phonon Property Calculation and Validation
Construct the dynamical matrix for wave vectors throughout the Brillouin zone through Fourier transformation of the real-space force constants:
[ D{\alpha\beta}(j,k;\mathbf{q}) = \frac{1}{\sqrt{mj mk}} \sumP \Phi{\alpha\beta}^{jk}(0,P) e^{-i\mathbf{q}\cdot[\mathbf{R}(P) + \mathbf{\tau}k - \mathbf{\tau}_j]} ]
where ( mj ) and ( mk ) are atomic masses, ( \mathbf{R}(P) ) is the lattice vector of cell ( P ), and ( \mathbf{\tau}_j ) is the basis vector for atom ( j ). Diagonalize the dynamical matrix to obtain phonon frequencies and eigenvectors. Validate the results by ensuring the acoustic sum rule is satisfied and checking for the absence of imaginary frequencies (soft modes) at high-symmetry points, unless physically justified by instabilities induced by the vacancies.
Table 2: Key Computational Parameters for Finite-Displacement Calculations
| Parameter | Typical Values | Impact on Accuracy |
|---|---|---|
| Displacement Magnitude | 0.01-0.03 Å | Too small: numerical noise; Too large: anharmonic effects |
| Supercell Size | 2×2×2 to 4×4×4 primitive cells | Larger cells reduce image interactions but increase computational cost |
| Energy Cutoff | 1.3-1.5× default cutoff for basis set | Higher values improve accuracy but increase computational cost |
| k-point Mesh | Γ-centered, density equivalent to 4×4×4 for primitive cell | Denser meshes improve accuracy, especially for metals |
| Force Convergence | 1-10 meV/Å for electronic self-consistency | Tighter criteria improve force constant accuracy |
Table 3: Research Reagent Solutions for Finite-Displacement Calculations
| Tool Category | Specific Software | Function and Application |
|---|---|---|
| DFT Electronic Structure | VASP [23], QUANTUM ESPRESSO [23], ABINIT [23] | Calculate forces and total energies for displaced structures |
| Phonon Calculation | Phonopy [23], ALAMODE [23], YPHON [23] | Implement finite-displacement method, extract force constants, calculate phonons |
| Supercell Generation | supercell program [21], ATAT [23] | Generate supercells with defects and enumerate configurations |
| Structure Manipulation | ASE, pymatgen | Structure visualization, manipulation, and workflow automation |
| Data Analysis | Python, NumPy, SciPy | Custom analysis scripts for force constant processing and phonon property calculation |
Advanced Considerations for Imperfect Lattices
Treatment of Long-Range Interactions in Polar Materials
For polar solids with vacancies, the mixed-space approach provides an effective solution for handling long-range dipole-dipole interactions [23]. This method treats the short-range interactions in real space while handling the long-range nonanalytic part in reciprocal space. The dynamical matrix is separated into analytic and nonanalytic components:
[ D{\alpha\beta}(j,k;\mathbf{q}) = D{\alpha\beta}^{\text{analytic}}(j,k;\mathbf{q}) + D_{\alpha\beta}^{\text{nonanalytic}}(j,k;\mathbf{q}) ]
The nonanalytic part accounts for the macroscopic electric field induced by atomic vibrations:
[ D{\alpha\beta}^{\text{nonanalytic}}(j,k;\mathbf{q}) = \frac{4\pi e^2}{\Omega} \frac{\sum\gamma q\gamma Z{\gamma\alpha}^(j) \sum_\delta q_\delta Z_{\delta\beta}^(k)}{\sum{\alpha,\beta} \epsilon{\alpha\beta} q\alpha q\beta} ]
where ( \Omega ) is the unit cell volume, ( Z^* ) is the Born effective charge tensor, and ( \epsilon_{\alpha\beta} ) is the electronic dielectric tensor. This approach correctly reproduces the LO-TO splitting in imperfect lattices, provided the Born effective charges and dielectric tensor are appropriately calculated for the defective supercell.
Convergence and Error Analysis
Systematic convergence tests are essential for reliable results. Key parameters requiring convergence analysis include:
- Supercell size: Test increasing supercell sizes until phonon frequencies and defect formation energies converge.
- k-point sampling: Ensure Brillouin zone integration is sufficiently accurate for force calculations.
- Displacement magnitude: Verify that results are stable with respect to small variations in displacement size.
- Energy and force convergence criteria: Tighten until phonon frequencies change by less than 0.1 THz.
Error analysis should account for both numerical errors from the DFT calculations and methodological limitations of the harmonic approximation, which becomes less reliable for systems with significant anharmonicity, such as those containing vacancies that may enhance atomic disorder.
Applications in Materials Research
The finite-displacement method for imperfect lattices enables the investigation of numerous structure-property relationships in defective materials:
- Thermal stability of defective materials: Predicting how vacancies influence free energy and phase stability at finite temperatures.
- Thermal conductivity tuning: Calculating how vacancy concentration and arrangement affect lattice thermal conductivity for thermoelectric applications.
- Defect identification: Using calculated phonon spectra as fingerprints to identify specific vacancy types in experimental vibrational spectroscopy.
- Ionic conductivity: Studying the coupling between phonons and ionic hopping mechanisms in superionic conductors with vacancy-mediated diffusion [24].
The following diagram illustrates the interconnected applications of this methodology in materials research:
Research Applications Diagram
The finite-displacement method provides a robust framework for extracting force constants and calculating phonon properties in imperfect lattices with vacancies. When implemented with careful attention to supercell construction, combinatorial configuration generation, and the treatment of long-range interactions in polar materials, this approach enables accurate prediction of thermal and vibrational properties of defective materials. The protocols outlined in this work establish a foundation for systematic investigations of vacancy impacts on materials behavior, with applications spanning from fundamental defect physics to the rational design of materials for energy technologies. As computational power increases and methods refine, the finite-displacement approach continues to offer valuable insights into the intricate relationship between atomic-scale defects and macroscopic materials properties.
In the study of crystalline materials, the concept of phonons—quantized lattice vibrations—provides a fundamental framework for understanding thermal, electrical, and mechanical properties. However, in real materials containing vacancies, defects, or compositional disorder, the perfect periodicity of the lattice is broken, leading to a phenomenon known as phonon broadening. This broadening manifests as finite phonon lifetimes and reduced phase coherence, critically affecting material properties from thermal conductivity to electronic behavior.
Within supercell lattice dynamics (SCLD), phonon broadening arises from the interaction between vibrational modes and disorder-induced scattering centers. The spectral function, a key theoretical construct, captures both the energy and lifetime of these excitations, providing a complete picture of how disorder modifies vibrational properties. This application note establishes standardized protocols for analyzing phonon broadening within SCLD frameworks, with particular emphasis on vacancy-type defects, enabling researchers to quantitatively predict and interpret disorder-induced effects in materials systems.
Core Principles and Theoretical Framework
Spectral Functions and Disorder-Induced Broadening
The phonon spectral function, A(q,ω), describes the probability of exciting a phonon with wavevector q and frequency ω. In perfectly ordered crystals, this function appears as a series of sharp delta functions. Disorder broadens these peaks, with the linewidth quantitatively relating to the phonon lifetime. The fundamental connection is given by:
Γ = ħ / τ
where Γ is the full width at half maximum (FWHM) of the spectral peak and τ is the phonon lifetime [25]. This broadening occurs because disorder opens new scattering channels that dissipate the vibrational energy and limit the phonon's spatial coherence.
In SCLD simulations, the spectral function can be computed through the phonon unfolding technique [10], which maps the vibrational modes of a large, disordered supercell back onto the Brillouin zone of a primitive cell. This approach directly captures the k-point-dependent broadening effects induced by disorder without relying on approximate averaging techniques.
Mass Disorder vs. Force-Constant Disorder
Two primary sources of phonon broadening exist in disordered systems:
- Mass disorder: Arises from atomic mass differences between constituent elements, primarily affecting acoustic modes at low frequencies.
- Force-constant disorder: Results from variations in chemical bonding environments, affecting phonons across the entire spectrum and often dominating in systems with minimal mass contrast [25].
Table 1: Characteristics of Phonon Broadening Mechanisms
| Disorder Type | Primary Effect | Dominant Phonon Modes | Key Experimental Signature |
|---|---|---|---|
| Mass Disorder | Scattering due to impedance mismatch | Low-frequency acoustic | Linewidth proportional to ω⁴ near Γ-point |
| Force-Constant Disorder | Scattering due to bond stiffness variations | All frequencies, particularly optical | Anisotropic broadening in different Brillouin zone directions |
| Vacancy Defects | Combined mass and force-constant effects | All frequencies | Enhanced broadening near specific q-points |
Recent research has challenged a long-standing assumption in mean-field theories, revealing that local chemical environments create significantly larger variations in species-pair-resolved force constants than previously accounted for in global averaging approaches [25]. This finding has profound implications for SCLD simulations, emphasizing the need for sufficiently large supercells to capture the true statistical distribution of local environments.
Quantitative Data on Phonon Broadening
Experimental Measurements in Model Alloys
Combined experimental and computational studies on equiatomic alloys provide crucial benchmarks for phonon broadening analysis. In NiCo and NiFe alloys—systems with minimal mass disorder but significant force-constant disorder—measurements reveal substantial phonon broadening, particularly at higher wavevectors.
Table 2: Experimental Phonon Linewidths in Disordered Alloys [25]
| Material | Phonon Mode | Wavevector (rlu) | FWHM Linewidth (meV) | Primary Broadening Cause |
|---|---|---|---|---|
| NiCo | TA₁ | [0.5, 0.5, 0] | ~1.2 | Force-constant disorder |
| NiCo | LA | [0.5, 0.5, 0] | ~0.8 | Force-constant disorder |
| NiFe | TA₁ | [0.5, 0.5, 0] | ~2.5 | Force-constant disorder |
| NiFe | LA | [0.5, 0.5, 0] | ~1.8 | Force-constant disorder |
Notably, NiFe exhibits significantly larger linewidths than NiCo across most of the Brillouin zone, with values remaining elevated (1.5-2.0 meV) even at wavevectors as low as 0.2 rlu [25]. This persistent broadening suggests strong scattering processes that cannot be explained by simple mass contrast models.
Electron-Phonon Coupling Contributions
In metallic systems, electron-phonon coupling (EPC) provides an additional broadening mechanism, with the contribution to linewidth described by:
γᴇᴘᴄ(q) = πω(q)∑ₖ|gₖ₊q,ₖ(q)|²δ(εₖ - εғ)δ(εₖ₊q - εғ)
where gₖ₊q,ₖ(q) represents the EPC matrix elements [26]. In materials like YNi₂B₂C, strong k-dependence of these matrix elements can cause significant phonon broadening even in the absence of Fermi surface nesting or lattice anharmonicity [26].
Diagram 1: SCLD Workflow for Phonon Broadening Analysis. The process begins with supercell construction and proceeds through force constant calculation to spectral analysis.
Experimental Protocols for SCLD with Vacancies
Supercell Construction and Vacancy Implementation
Objective: Create structurally representative supercells with controlled vacancy concentrations for phonon property calculation.
Materials and Computational Methods:
- DFT code with phonon capabilities (VASP, Phonopy, ABINIT, Quantum ESPRESSO)
- Structure visualization software (VESTA, VMD)
- Spectral unfolding code (SCUILD, UNFOLD, PHONON-UNFOLDING)
Procedure:
- Primitive Cell Optimization: Begin with fully relaxed primitive cell of the perfect crystal using DFT with convergence criteria of at least 10⁻⁸ eV for electronic energy and 10⁻² eV/Å for forces [27].
Supercell Generation: Create a 3×3×3 or larger supercell expansion to ensure vacancy separation distances exceed the force constant cutoff radius. For systematic studies, generate supercells with dimensions 2×2×2, 3×3×3, and 4×4×4 to test for convergence.
Vacancy Introduction:
- Remove one atom from the supercell to create a single vacancy (N-1 atoms).
- For controlled concentration studies, remove multiple atoms while maintaining minimum separation distances.
- For random distributions, employ special quasirandom structures (SQS) for statistically representative sampling.
Supercell Relaxation: Perform full ionic relaxation of the defective supercell using the same convergence criteria as step 1. This allows lattice distortion around the vacancy to be fully captured.
Critical Considerations:
- Vacancy concentration should typically not exceed 5% to maintain physical relevance and avoid supercell-size artifacts.
- The supercell must be sufficiently large to prevent spurious vacancy-vacancy interactions through periodic boundary conditions.
- For charged vacancies, include appropriate background charge compensation in DFT calculations.
Force Constant Calculation and Spectral Unfolding
Objective: Compute the force constant matrix and extract the phonon spectral function with broadening information.
Procedure:
- Force Constant Extraction:
- Use the finite-displacement method as implemented in Phonopy [27] or similar packages.
- Apply displacements of 0.01-0.03 Å to each independent atom in the supercell.
- Calculate the Hellmann-Feynman forces for each displaced configuration.
Dynamical Matrix Construction:
- Build the mass-weighted dynamical matrix from the force constants.
- Diagonalize the dynamical matrix to obtain eigenvalues (squared frequencies) and eigenvectors (phonon modes).
Spectral Unfolding:
- Apply the unfolding technique to map supercell phonons to the primitive Brillouin zone [10].
- Calculate the spectral function using the projection operator:
A(q,ω) = ∑j |
Linewidth Extraction:
- At each q-point, fit the spectral function with Lorentzian profiles.
- Extract the FWHM as the phonon linewidth Γ(q).
- Convert to phonon lifetime using τ(q) = ħ/Γ(q).
Validation Steps:
- Compare phonon frequencies of perfect supercell with primitive cell results to verify implementation.
- Ensure the integrated spectral weight is conserved through the unfolding process.
- Check that acoustic sum rules are satisfied in the force constant matrix.
Analysis of Vacancy-Induced Broadening
Objective: Quantify and interpret vacancy-induced phonon broadening across the Brillouin zone.
Procedure:
- q-Path Selection: Choose a high-symmetry path through the Brillouin zone (e.g., Γ-X-K-Γ) for detailed analysis.
Mode-Projected Analysis: Decompose the total broadening into contributions from different phonon branches (acoustic vs. optical, longitudinal vs. transverse).
Spatial Localization: Calculate the participation ratio of each mode to identify localized vibrations near vacancy sites: PR = 1/(N∑i ei⁴) where e_i is the displacement amplitude of atom i.
Force-Constant Analysis: Compare the force constants in the vicinity of the vacancy with bulk values to quantify the local bonding perturbation.
Thermal Conductivity Prediction: Use the extracted lifetimes in the Boltzmann Transport Equation to predict the reduction in lattice thermal conductivity: κ = 1/3∑q vg(q)² C(q) τ(q) where v_g is the group velocity, C is the heat capacity, and τ is the lifetime.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Table 3: Key Computational Tools for SCLD Phonon Broadening Analysis
| Tool/Software | Primary Function | Application in Phonon Broadening | Key Features |
|---|---|---|---|
| VASP | DFT Electronic Structure | Force constant calculation | PAW pseudopotentials, phonon module |
| Phonopy | Phonon Analysis | Force constant extraction & diagonalization | Finite displacement method, supercell support |
| Phono3py | Three-Phonon Scattering | Anharmonic contributions to broadening | Third-order force constants, lifetime calculation |
| ALAMODE | Anharmonic Lattice Dynamics | Higher-order phonon scattering | Compressive sensing for force constants |
| ShengBTE | Boltzmann Transport | Thermal conductivity from lifetimes | Iterative solution, impurity scattering |
| UNFOLD | Spectral Unfolding | Phonon spectral functions | Supercell-to-primitive mapping, broadening visualization |
Visualization and Data Interpretation Protocols
Diagram 2: Data Integration for Phonon Broadening Interpretation. Comparative analysis between experimental and theoretical approaches enables robust model validation.
The protocols established in this application note provide a comprehensive framework for analyzing phonon broadening within supercell lattice dynamics, with specific application to vacancy-containing systems. The integration of spectral unfolding techniques with ab initio force constant calculations enables quantitative prediction of disorder-induced phonon lifetime effects, bridging the gap between idealized crystal models and real material systems.
As computational capabilities expand, future methodologies will likely incorporate higher-order anharmonic effects [27], temperature-dependent force constants, and more sophisticated treatments of electron-phonon coupling [26]. The ongoing development of efficient spectral unfolding algorithms and machine learning accelerated force field generation promises to extend these analyses to larger supercells and more complex defect structures, further enhancing our understanding of phonon broadening phenomena in functional materials.
In the context of supercell lattice dynamics (SCLD) for phonon calculations with vacancies, the band unfolding technique is an essential post-processing tool for interpreting complex computational results. When a supercell containing a vacancy or defect is created, its Brillouin Zone (BZ) becomes smaller than that of the primitive cell, causing electronic bands and phonon dispersions to "fold" into this reduced reciprocal space. This folding obscures the direct relationship between the defective system's properties and the well-understood band structure of the pristine material. Band unfolding effectively reverses this folding process, mapping the supercell's spectral information back into the primitive BZ, thereby producing an effective band structure that researchers can intuitively relate to the pristine system's characteristics.
The fundamental challenge addressed by band unfolding arises from the mathematical construction of supercells. As defined by the transformation matrix M that relates the supercell lattice vectors (A) to the primitive cell vectors (a) through A = M · a, the number of primitive cells in the supercell equals det(M) = Nc [28]. This transformation causes Nc distinct k-points from the primitive Brillouin zone (PBZ) to fold onto a single K-point in the supercell Brillouin zone (SBZ), following the relation k = K + G, where G is a reciprocal lattice vector of the supercell [29]. For SCLD investigations of vacancies, this folding mechanism complicates the direct interpretation of how point defects alter vibrational and electronic properties, making unfolding techniques indispensable for meaningful analysis.
Theoretical Foundation of Band Unfolding
Mathematical Formalism
The core mathematical operation in band unfolding involves calculating the spectral function A(k,ε), which represents the probability of finding a state with crystal momentum k in the primitive cell and energy ε in the supercell calculation. This spectral function is computed as:
[A(\vec{k},\epsilon) = \summ P{\vec{K}m}(\vec{k}) \delta(\epsilon_{\vec{K}m}-\epsilon)]
where (P_{\vec{K}m}(\vec{k})) are the unfolding weights that quantify how much of the character of the primitive cell state |kn⟩ is preserved in the supercell state |Km⟩ [29]. These weights can be computed without explicit knowledge of the primitive cell eigenstates through the formula:
[P{\vec{K}m}(\vec{k}) = \sum{sub{\vec{G}}} |C_{\vec{K}m}(\vec{G}+\vec{k}-\vec{K})|^2]
where (C_{\vec{K}m}) are the Fourier coefficients of the supercell eigenstate |Km⟩ and the summation is over a specific subset of reciprocal space vectors of the supercell that match the reciprocal space vectors of the primitive cell [29].
An alternative approach implemented in the KPROJ code uses the k-projection method, which builds a projector operator based on translation operators and their irreducible representations labeled by k within the first BZ of the primitive cell [28]. This method leverages the transformation matrix M to systematically connect the supercell and primitive cell representations.
Unfolding Workflow Logic
The following diagram illustrates the logical workflow of band unfolding from supercell to primitive Brillouin zone:
Computational Implementation and Software Tools
Available Band Unfolding Codes
Table 1: Comparison of band unfolding software packages
| Software | Implementation | Supported Codes | Key Features | Applicability to SCLD |
|---|---|---|---|---|
| easyunfold | Python | VASP | Symmetry-breaking accounting, k-point sampling automation | Electronic structure analysis of defective systems [30] |
| GPAW Unfold | Python | GPAW (LCAO, PW, real-space) | Real-space unfolding method | Defect states identification (e.g., MoS₂ with S vacancy) [29] |
| KPROJ | Fortran 90 | VASP, QE, ABINIT, ABACUS, PHONOPY | k-projection method, layer projection, phonon unfolding | Direct phonon band unfolding, interface systems [28] |
| Siesta Unfold | Standalone | Siesta | Full unfolding and refolding to arbitrary BZ | Amorphous materials, vacancy studies in bulk and 2D materials [31] |
The Scientist's Toolkit: Essential Research Reagents
Table 2: Essential computational tools for supercell band unfolding
| Tool Category | Specific Package/Function | Purpose in Workflow |
|---|---|---|
| DFT Codes | VASP, Quantum ESPRESSO, ABINIT, GPAW, Siesta | Perform initial supercell ground state calculations [29] [28] [31] |
| Unfolding Software | easyunfold, KPROJ, GPAW.unfold, Siesta/Unfold | Post-process wavefunctions to generate unfolded bands [30] [29] [28] |
| Structure Generation | supercell program | Generate disordered supercells with vacancies and substitutions [21] |
| Transformation Matrix | Matrix M relating primitive and supercell | Defines the folding relationship between BZs [29] [28] |
| Spectral Function Analysis | Weight calculation and interpolation | Quantifies primitive character in supercell states [29] [28] |
Experimental Protocol: Band Unfolding for Defective Systems
The initial step involves creating an appropriate supercell structure containing the vacancy defect:
Primitive Cell Definition: Begin with the optimized primitive unit cell of the pristine material. For example, in the case of silicon, this would be the conventional diamond cubic cell with lattice parameter ~5.430 Å [31].
Supercell Generation: Apply the transformation matrix M to create the supercell. For instance, a 3×3×1 supercell for 2H-MoS₂ would use M = [[3,0,0],[0,3,0],[0,0,1]] [29]. The supercell program provides a systematic approach for generating such structures, implementing combinatorial algorithms to handle atomic substitutions and vacancies while considering symmetry-equivalent configurations [21].
Defect Introduction: Introduce the vacancy by removing the appropriate atom(s). For example, in the MoS₂ case, a single sulfur atom is deleted from the structure [29]. In silicon, one might remove a single atom from an 8-atom supercell to study monovacancies [31].
Structure Relaxation: Perform a full geometry optimization of the defective supercell using DFT to obtain the realistic atomic positions in the presence of the vacancy.
Ground State and Band Structure Calculations
The computational workflow for the unfolding procedure consists of:
Ground State Calculation: Perform a self-consistent field (SCF) calculation for the defective supercell. For example, using GPAW with LCAO mode ('dzp' basis) for MoS₂, with appropriate k-point sampling (e.g., 4×4×1), and Fermi-Dirac smearing (e.g., 0.01 eV) [29]. Critical output files containing wavefunctions must be saved (e.g., .gpw in GPAW, .HSX in Siesta, WAVECAR in VASP).
k-path Definition: Define the desired high-symmetry path in the primitive Brillouin zone (e.g., M-K-Γ for hexagonal systems) [29].
k-point Mapping: For each k-point in the primitive BZ path, find the corresponding K-point in the supercell BZ using the transformation matrix M and the relation k = K + G [29] [28].
Non-Self-Consistent Calculation: Perform a non-SCF calculation at the mapped K-points to obtain eigenvalues and eigenvectors across the desired energy range.
Unfolding Execution and Analysis
Unfolding Setup: Initialize the unfolding object with the appropriate parameters, including the transformation matrix M and the wavefunction file [29].
Spectral Function Calculation: Compute the spectral function A(k,ε) using the implemented unfolding method (e.g., k-projection in KPROJ [28], real-space method in GPAW [29], or refolding approach in Siesta [31]).
Result Visualization: Plot the spectral function along the high-symmetry path, often representing the intensity with a color scale. Defect-induced states, such as gap states from vacancies, appear as additional features compared to the pristine band structure [29].
Case Study: Sulfur Vacancy in MoS₂
Application of the Unfolding Protocol
A practical example demonstrates the unfolding procedure for a sulfur vacancy in MoS₂:
System Preparation: Create a 3×3×1 supercell of 2H-MoS₂ and remove one sulfur atom [29].
Calculation Parameters: Use GPAW in LCAO mode with DZP basis set, LDA functional, and 4×4×1 k-point sampling for the ground state calculation [29].
k-path Selection: Define the path M-K-Γ in the hexagonal primitive Brillouin zone with 48 points along the path [29].
Unfolding Execution: Employ GPAW's unfolding module to calculate the spectral function, which clearly reveals defect states within the band gap that result from the sulfur vacancy [29].
This application highlights how unfolding makes defect states visible and interpretable within the familiar context of the primitive Brillouin zone, enabling direct comparison with angle-resolved photoemission spectroscopy (ARPES) experiments [28].
Advanced Applications in Lattice Dynamics
Phonon Band Unfolding
While primarily developed for electronic structure, band unfolding techniques have been extended to phonon systems, particularly relevant for SCLD with vacancies:
Phonon Unfolding: The KPROJ program explicitly supports unfolding of phonon bands, enabling the study of how vacancies affect vibrational spectra [28].
Methodology: Phonon unfolding employs similar mathematical foundations, projecting the vibrational modes of the supercell onto the primitive Brillouin zone to understand how defects alter phonon dispersions.
Spin-Lattice Dynamics: Advanced SCLD simulations that couple spin and lattice degrees of freedom, such as studies of vacancy formation and migration in ferromagnetic BCC iron, can benefit from unfolding to interpret complex magnetic and vibrational interactions [32].
Layer-Projected Unfolding
For surface and interface systems, KPROJ implements a specialized layer projection technique that accelerates the calculation using a combination of Fast Fourier Transform (FFT) and inverse FFT algorithms [28]. This allows researchers to obtain effective unfolded band structures for specific layers in heterogeneous systems, which is particularly valuable for studying vacancy effects at surfaces or interfaces.
Band unfolding techniques represent a crucial methodology for bridging the gap between supercell calculations containing defects and the intuitive interpretation of results within the primitive Brillouin zone framework. For researchers investigating vacancies using supercell lattice dynamics, these methods provide an essential tool for connecting computational predictions with experimental observations, particularly in interpreting how point defects alter electronic, vibrational, and magnetic properties. The availability of multiple well-documented software implementations ensures that scientists can select the approach best suited to their specific computational framework and research objectives, advancing our understanding of defect engineering in materials design.
The independent control of thermal and electrical properties is a paramount objective in the development of advanced materials for energy conversion and electronic applications [33]. In thermoelectric materials, for instance, a low thermal conductivity (κ) must be combined with high electrical conductivity to achieve a high figure of merit, ZT [33]. Phonons, the primary heat carriers in semiconductors and insulators, are significantly scattered by atomic-scale defects, making vacancy engineering a powerful strategy for thermal conductivity reduction. This case study explores the application of Supercell Lattice Dynamics (SCLD) to predict how engineered vacancies can drastically suppress thermal transport, using insights from recent first-principles studies and molecular dynamics simulations. The core thesis is that SCLD provides a critical computational framework for understanding vacancy-phonon interactions, enabling the rational design of materials with tailored thermal properties.
The foundational principle is that vacancies disrupt the perfect periodicity of a crystal lattice, leading to phonon scattering through two primary mechanisms:
- Mass-difference scattering due to the change in local atomic mass.
- Strain-field scattering caused by the distortion of bonds around the vacancy site [34] [35]. These effects are quantitatively captured by SCLD calculations, which model the modified interatomic force constants in a supercell containing a defect, allowing for the computation of perturbed phonon frequencies and scattering rates.
Theoretical and Computational Foundation
The Role of Vacancies in Thermal Conductivity Reduction
The introduction of vacancies into a crystal lattice creates scattering centers that dissipate phonon momentum, thereby reducing lattice thermal conductivity (κL). The efficacy of this scattering is concentration-dependent, as demonstrated in a molecular dynamics study on CH₃NH₃PbI₃. The results showed that the lattice thermal conductivity generally decreases as the vacancy concentration increases from 0% to 1%. This reduction was correlated with a decrease in the material's sound velocity, a parameter directly accessible from SCLD-derived phonon dispersions [35].
Beyond simple point scattering, vacancies can induce complex structural changes that further impact phonon transport. In titanium monoxide (TiO), the presence of ~12.5% vacancy concentration was found to eliminate imaginary phonon modes in the cubic phase, dynamically stabilizing a structure that is otherwise unstable at room temperature. This dramatic alteration of the phonon dispersion spectrum has profound implications for thermal transport properties [34].
SCLD as a Predictive Tool
SCLD extends standard lattice dynamics by employing a large, defect-containing supercell to compute the force constants and phonon spectrum of the imperfect lattice. This method allows researchers to:
- Quantify phonon lifetime reduction by calculating the scattering rates of phonons due to vacancies.
- Model the impact of vacancy concentration by varying the defect density within the supercell.
- Predict thermal conductivity by integrating the modified phonon properties into the solution of the Boltzmann Transport Equation (BTE) or by using the relaxation time approximation.
A key advantage of SCLD is its ability to probe the wave nature of phonons and related localization effects. In disordered twisted multilayer graphene, strong phonon localization—a wave interference phenomenon—was identified as the mechanism behind a giant (up to 80%) reduction in cross-plane thermal conductivity [36]. While not a vacancy system, this underscores the importance of modeling coherent phonon effects, for which SCLD is well-suited.
Key Evidence from the Literature
The following table summarizes quantitative findings on vacancy-induced thermal conductivity reduction from recent studies, providing a benchmark for SCLD predictions.
Table 1: Experimental and Computational Evidence of Vacancy-Induced Thermal Conductivity Reduction
| Material System | Vacancy Type & Concentration | Thermal Conductivity Reduction | Key Mechanism | Methodology |
|---|---|---|---|---|
| CH₃NH₃PbI₃ [35] | Vₘₐ, VPb, VI (up to 1%) | Overall decrease with concentration, with some slight variations | Reduced sound velocity and phonon scattering | Classical Molecular Dynamics |
| Cubic TiO [34] | Ti and O vacancies (~12.5%) | System dynamically stabilized; low κ inferred | Elimination of imaginary phonon modes, altered phonon dispersion | Density Functional Theory (Phonon Calculations) |
| Connected Si Nanodots [33] | Interfaces acting as vacancy-like scatterers | Below/close to the amorphous limit | Phonon scattering at nanodot interfaces | Not specified (Review of experimental structures) |
Furthermore, the formation energy of vacancies, a critical parameter determining their equilibrium concentration, is highly sensitive to the local chemical environment. In complex materials like entropy-stabilized oxides (ESOs), the vacancy formation energy is not a single value but a distribution. For example, first-principles calculations revealed that the oxygen vacancy formation energy in (Mg₀.₂Ni₀.₂Co₀.₂Cu₀.₂Zn₀.₂)O can range from 0.17 eV to 2.54 eV depending on the local cation configuration [37]. This highlights the necessity of using a supercell approach to capture the full spectrum of defect behavior in disordered systems.
SCLD Application Protocol: Predicting Vacancy Effects in TiO
This protocol outlines the application of SCLD to investigate the phonon properties and thermal conductivity reduction in titanium monoxide (TiO) with vacancies, based on the study by Hosseini et al. [34].
The following diagram illustrates the integrated computational workflow for an SCLD study, from supercell construction to thermal property prediction.
Step-by-Step Methodology
Step 1: Supercell Construction and Structural Relaxation
- Procedure: Build a supercell of the pristine crystal structure (e.g., cubic or monoclinic TiO). Introduce vacancies at specific lattice sites, varying concentrations systematically (e.g., 0%, ~6.25%, ~12.5%). Perform a full geometry optimization using Density Functional Theory (DFT) to relax the ionic positions and cell volume. This step accounts for the local structural distortions around the vacancy.
- Computational Details: Use a plane-wave basis set code like VASP. Employ the Projector Augmented-Wave (PAW) method and a suitable exchange-correlation functional (e.g., PBE). Set an energy cutoff of ≥ 450 eV and k-point mesh to ensure total energy convergence of < 1 meV/atom [38] [34].
Step 2: Force Constant Calculation
- Procedure: In the relaxed, vacancy-containing supercell, compute the second-order interatomic force constants (IFCs). This is done by calculating the Hellmann-Feynman forces in response to small finite displacements of atoms from their equilibrium positions. The force constants represent the "springs" between atoms in the lattice.
- Details: The finite displacement method is typically used. A displacement of ~0.01 Å is standard. The force constant matrix will clearly show the broken bonds and modified interactions around the vacancy site.
Step 3: Phonon Dispersion and Density of States Calculation
- Procedure: Diagonalize the dynamical matrix constructed from the force constants to obtain the phonon frequencies and eigenvectors across the Brillouin zone. This yields the phonon dispersion curves and the phonon Density of States (DOS).
- Analysis: Compare the results with the pristine cell. Key observations may include:
- The removal of high-frequency optical modes associated with the missing atom.
- Shifts in the frequencies of remaining modes due to strain.
- The appearance of localized modes or resonances, if any.
- As shown in TiO, the stabilization of the cubic phase by removing imaginary modes (soft modes) at the Γ-point [34].
Step 4: Phonon Scattering Rate and Thermal Conductivity Calculation
- Procedure: Implement a perturbation theory approach (e.g., the relaxation time approximation, RTA) to calculate the phonon-vacancy scattering rates. The scattering rate for a phonon mode λ is given by ( \frac{1}{τ{λ}} = n{vac} \cdot Γ{λ} \cdot ω{λ}^{2} ), where ( n{vac} ) is the vacancy concentration, ( Γ{λ} ) is a mode-dependent scattering strength, and ( ω{λ} ) is the phonon frequency. The lattice thermal conductivity (κL) is then computed by solving the BTE: [ κL = \frac{1}{NV} \sumλ Cλ vλ^2 τλ ] where *N* is the number of q-points, *V* is volume, *Cλ* is the mode heat capacity, and v_λ is the phonon group velocity.
- Validation: Compare the calculated κ_L for the pristine system with known experimental or theoretical values to validate the computational protocol.
Table 2: Key Computational Tools and Resources for SCLD Studies
| Resource / Software | Type | Function in SCLD/Vacancy Studies |
|---|---|---|
| VASP [38] [34] | Software Package | Performs DFT calculations for structural relaxation, electronic structure, and force calculations in supercells. |
| Phonopy | Software Package | Post-processes force constants from DFT to calculate phonon dispersion, DOS, and thermal properties. |
| GPUMD [36] | Software Package | Performs highly efficient molecular dynamics simulations, including thermal conductivity calculation using NEMD. |
| PAW-PBE Pseudopotentials [38] | Computational Parameter | Standard, efficient pseudopotentials used in DFT to represent core electrons and their interaction with valence electrons. |
| HSE06/SCAN Functionals [38] | Computational Parameter | Advanced exchange-correlation functionals that provide more accurate defect formation energies and electronic structures than standard PBE. |
| Bayesian Optimization/CNN [36] | Algorithm | Machine learning methods used to efficiently navigate vast configuration spaces (e.g., defect positions, alloy compositions) to find structures with minimal thermal conductivity. |
This case study demonstrates that SCLD is an indispensable tool for advancing vacancy engineering in materials science. By moving beyond empirical approaches, SCLD provides a fundamental, physics-based understanding of how vacancies alter phonon spectra, induce localization, and ultimately suppress thermal transport. The protocol outlined for TiO offers a generalizable template that can be adapted to other material systems, from perovskites for photovoltaics to complex high-entropy oxides. The integration of SCLD with high-throughput computations and machine learning, as previewed in other contexts [37] [36], represents the future frontier for the rational, computationally driven design of materials with ultralow and precisely tuned thermal conductivity.
Overcoming Computational Challenges: Optimizing SCLD for Complex Vacancy Systems
Supercell lattice dynamics (SCLD) is a foundational technique for investigating phonon properties in materials with point defects, such as vacancies. However, the computational cost associated with constructing and simulating large supercells often becomes a significant bottleneck. Traditional approaches require supercells large enough to prevent spurious interactions between periodic images of defects, leading to system sizes that can be prohibitively expensive for direct ab initio calculations. This application note outlines structured protocols and advanced sampling strategies to optimize supercell sizing and sampling, enabling efficient and accurate SCLD simulations within phonon and vacancy research.
Foundational Concepts and Key Challenges
The core challenge in SCLD is to balance computational feasibility with physical accuracy. Two primary sources of computational expense are:
- Supercell Size for Defect Isolation: Modeling a single, isolated defect often requires a large supercell to minimize the elastic and electrostatic interactions between the defect and its periodic images. The required size can be substantial for accurate phonon property calculations.
- Configuration Space Sampling: For high defect concentrations or complex solid solutions, a vast number of distinct atomic configurations are possible. An exhaustive search for the most stable or representative configurations via density functional theory (DFT) is frequently intractable [21].
Addressing these challenges requires a move beyond brute-force computation towards smarter, more efficient algorithmic and sampling strategies.
Quantitative Comparison of Sampling and Sizing Methods
The table below summarizes the quantitative benefits and typical use cases of different modern approaches for mitigating computational costs in supercell-based studies.
Table 1: Comparison of Methods for Efficient Supercell Sizing and Sampling
| Method Name | Key Performance Metric | Reported Efficiency Gain | Primary Application Context |
|---|---|---|---|
| Small-Cell Sampling (SCS) [39] | Uses small, 1-2 element cells for training | Bypasses iterative active learning; avoids large-cell DFT | Multi-principal element alloys (MPEAs) |
| Machine Learning Potentials (MLPs) [40] | Reduction in CPU time for SSCHA | ~96% cost reduction for PdCuH2 [40] | Anharmonicity and quantum effects |
| Adaptive Basis Set Algorithm [41] | Eliminates need for large supercells | Retains reliable results with reduced cost | Mismatched material interfaces |
| Exhaustive Combinatorial Search [21] | Generates all symmetry-inequivalent structures | Handles complex disorder systematically | Substitutional defects and vacancies |
| Special Quasirandom Structures (SQS) [42] | Generates a few representative supercells | Aims to mimic random distribution | Metallic alloys, semiconductors |
Detailed Experimental Protocols
Protocol 1: High-Throughput Screening with Lattice Dynamics Descriptors
This protocol leverages machine learning (ML) to rapidly identify materials with desirable phonon transport properties, as demonstrated for sodium superionic conductors [24].
Initial Dataset Curation:
- Source: Extract structures from databases like the Open Quantum Materials Database (OQMD) and the Inorganic Crystal Structure Database (ICSD).
- Filtering: Apply criteria such as a non-zero band gap and negative formation energy to select thermodynamically plausible structures. The foundational study started with 3903 Na-containing structures [24].
Structure Re-optimization and Dynamic Stability Check:
- Re-optimize all filtered structures using DFT with strict parameters.
- Screen for dynamic stability by calculating phonon dispersion and excluding structures with imaginary frequencies. In the referenced workflow, 921 of 3903 initial structures passed this step [24].
Calculation of Lattice Dynamics Descriptors: For the dynamically stable structures, compute key phonon-derived descriptors using ML potentials (e.g., a fine-tuned EquiformerV2 model [24]) or DFT. Essential descriptors include:
- Phonon mean squared displacement (MSD) of the mobile ion.
- Acoustic cutoff frequency.
- Center of the vibrational density of states (VDOS) for the mobile ion.
- Degree of low-frequency vibrational coupling between the mobile ion and the host lattice.
Machine Learning Model Training and Prediction:
- Train an ML model (e.g., a deep neural network) using the calculated descriptors as input to predict the target property, such as diffusion coefficient or ionic conductivity.
- Use the trained model to rapidly screen vast material spaces, identifying candidate materials with predicted high performance.
Protocol 2: Efficient Supercell Configuration Sampling for Solid Solutions
This protocol is designed for systematically exploring local cation ordering in solid solutions, as applied to (Fe,Mg)₂SiO₄ olivine and (Fe,Mg)O ferropericlase [43].
Supercell Generation:
- Start from the pure end-member crystal structure (e.g., Mg₂SiO₄).
- Generate a supercell of sufficient size (e.g., 10×10×2 for olivine, resulting in ~5600 atoms) to model the target composition (e.g., Fe₀.₅Mg₀.₅) and capture relevant local interactions [43].
Introduction of Chemical Disorder:
- Use a program like
DISCUSto perform a two-step process [43]:- Random Substitution: Randomly substitute atoms (e.g., Mg with Fe) to achieve the target overall composition.
- Monte Carlo Optimization: Perform Monte Carlo simulations that swap atoms to achieve a target short-range order (SRO) parameter, α. Generate configurations representing different limiting cases:
- Clustering (α > 0): Favors like-atom pairing.
- Random (α ≈ 0): No preference.
- Anti-clustering (α < 0): Favors hetero-atom pairing.
- Use a program like
Property Calculation and Analysis:
- Structurally relax each generated supercell configuration using an appropriate force field or DFT.
- Calculate the target physical properties (e.g., elastic moduli, phonon dispersion, heat capacity) for each configuration.
- Analyze the results to determine the influence of the specific local cation arrangement versus the overall composition on the material's properties.
Protocol 3: Integrating Machine Learning Potentials with Quantum Methods
This protocol combines MLPs with the Stochastic Self-Consistent Harmonic Approximation (SSCHA) to efficiently model strong anharmonicity and quantum nuclear effects, as validated on PdCuH₂ [40].
Initial Small-Supercell SSCHA:
- Begin with a small supercell (e.g., the primitive unit cell).
- Generate an initial set of displaced atomic configurations and compute their energies, forces, and stresses (EFS) using DFT.
- Use these EFS data to train an initial Machine Learning Interatomic Potential (MTP).
Active Learning Cycle:
- For subsequent SSCHA populations, use the current MTP to predict the EFS of new configurations.
- Identify configurations where the MTP is uncertain (using an extrapolation grade γ). Compute these "extrapolative" configurations with DFT and add them to the training set.
- Retrain the MTP with the updated dataset.
- Repeat until the SSCHA calculation converges for the current supercell size.
Progressive Upscaling:
- Once converged for the small cell, upscale to a larger supercell (e.g., 2×2×2, then 3×3×3).
- Use the final MTP from the smaller cell as the initial potential for the larger cell's SSCHA, repeating the active learning cycle.
- This leverages the fact that most atomic interactions are captured in smaller cells, drastically reducing the number of required DFT calculations at larger supercell sizes. This workflow achieved a 96% reduction in CPU time for PdCuH₂ [40].
Workflow Visualization: Integrated SCLD Framework
The following diagram illustrates the synergistic relationship between the sizing, sampling, and computational methods described in the protocols.
The Scientist's Toolkit: Essential Research Reagents and Software
Table 2: Key Software and Computational Tools for Efficient SCLD
| Tool Name | Type / Category | Primary Function in Research |
|---|---|---|
| supercell [42] [21] | Standalone Program | Generates derivative supercell structures for atomic substitutions/vacancies; handles combinatorics, charge balancing, and filters symmetry-equivalent structures. |
| DISCUS [43] | Simulation Software | Introduces and refines chemical short-range order in supercells via Monte Carlo simulations. |
| Machine Learning Interatomic Potentials (MLIP) [40] | Code Package / Method | Creates fast, accurate surrogate potentials trained on DFT data to replace expensive direct DFT in large-scale MD or phonon calculations. |
| SSCHA [40] | Computational Method | Calculates vibrational properties, accounting for strong anharmonicity and quantum nuclear effects. |
| EquiformerV2 [24] | Machine Learning Potential | A universal ML potential that can be fine-tuned for specific materials systems to predict lattice dynamics properties and perform molecular dynamics simulations. |
| SQS Approach [42] | Modeling Methodology | Generates a minimal number of supercell configurations that best mimic the correlation functions of a perfectly random alloy. |
The accurate and efficient calculation of phonon properties is a cornerstone of modern computational materials science, essential for understanding thermal conductivity, phase stability, and dynamical behavior. Supercell lattice dynamics (SCLD) provides a rigorous framework for these calculations but traditionally requires computationally intensive density functional theory (DFT) calculations on multiple displaced supercells. The emergence of universal machine learning interatomic potentials (MLIPs) represents a paradigm shift, offering the potential to accelerate these calculations by orders of magnitude while maintaining near-DFT accuracy. These potentials, trained on extensive datasets spanning diverse chemical spaces, can serve as drop-in replacements for DFT in SCLD workflows, dramatically increasing throughput for screening materials and investigating complex defective systems, including those with vacancies. This Application Note outlines the practical deployment of universal MLIPs, evaluating their performance, detailing integration protocols into SCLD workflows, and providing a benchmarking case study for phonon calculations in materials with point defects.
Performance Evaluation of Universal MLIPs
Comprehensive benchmarking is crucial for selecting an appropriate universal MLIP. Performance varies significantly across models, especially for predicting finite-temperature dynamic properties and handling distorted atomic configurations common in defective systems.
Table 1: Benchmarking Universal MLIPs for Phonon and Defect Calculations
| Model | Phonon Frequency MAE (THz) | Dynamical Stability Classification Accuracy | Performance on Point Defects | Notable Strengths & Weaknesses |
|---|---|---|---|---|
| MACE-MP-0 | ~0.18 (on specialized models) [44] | High [44] | Reliable for defect phonon modes [45] | High accuracy, lower failure rate in relaxations [46] |
| CHGNet | Low [46] | High [46] | Good performance on defect structures [45] | Robust, low relaxation failure rate [46] |
| Mattersim-v1 | Low [46] | High [46] | Best for photoluminescence spectra of defects [45] | Top performer for phonons in pristine crystals [45] |
| M3GNet | Moderate [47] [46] | Moderate [47] | Used in high-throughput screening workflows [24] | Pioneer model; shows instability in some phonon spectra [47] |
| eqV2-M | Low [46] | High [46] | Not the top performer [45] | High phonon accuracy; higher relaxation failure rate [46] |
| ORB | Inconsistent [47] [46] | Low [47] | Not the top performer [45] | Can exhibit unphysical instabilities in MD [47] |
| SevenNet-0 | Low [47] [46] | High [47] | Good performance on defect structures [45] | Accurate phonon spectra [47] |
Universal MLIPs trained on Perdew-Burke-Ernzerhof (PBE) data can inherit the functional's biases, such as overestimating the tetragonality ((c/a) ratio) in a material like PbTiO₃ [47]. This can lead to failures in simulating realistic finite-temperature phase transitions under molecular dynamics (MD) [47]. Furthermore, models that predict forces as a separate output, rather than as derivatives of the energy (e.g., ORB, eqV2-M), can exhibit high failure rates in geometry optimizations due to inconsistent forces and energies [46] [45].
Integrated Workflow for SCLD with MLIPs
The following workflow diagram summarizes the protocol for performing phonon calculations of systems with vacancies using universal MLIPs, from initial structure preparation to final analysis.
The diagram outlines the key stages of a supercell lattice dynamics calculation for a system with vacancies, leveraging universal MLIPs for acceleration. The process begins with a primitive cell definition, proceeds through supercell construction and defect configuration sampling, and uses the MLIP for the computationally intensive steps of relaxation and force calculation before final phonon analysis.
Experimental Protocols
Protocol 1: High-Throughput Phonon Calculation with MACE
This protocol, adapted from Lee & Xia (2024), uses a reduced number of supercells to train a specialized MACE model for rapid phonon calculations [44].
- Structure Selection & Preparation:
- Select a diverse set of crystal structures for the target chemical space.
- For each material, generate a
2x2x2or3x3x3supercell using a tool likephonopy.
- Training Data Generation:
- For each supercell, create 6-8 structurally distinct configurations by applying random displacements of 0.01–0.05 Å to all atoms. This contrasts with the finite-displacement method, which displaces one atom at a time.
- Perform DFT single-point calculations on these displaced supercells to obtain the total energy and atomic forces for each configuration. This constitutes the training dataset.
- Model Training:
- Train a MACE model on the aggregated dataset from all materials.
- Key Hyperparameters: A 4-body MPNN scheme, a radial cutoff of 5.0 Å, and data shuffling across all materials to maximize model generalizability.
- Phonon Calculation & Validation:
- For a new material of interest, use the trained MACE model to compute the forces for a set of systematically displaced supercells (e.g., using the
phonopypackage). - The model-predicted forces are used to construct the dynamical matrix and compute the harmonic phonon frequencies and density of states.
- Validate the model predictions against a small subset of materials calculated with full DFT phonons.
- For a new material of interest, use the trained MACE model to compute the forces for a set of systematically displaced supercells (e.g., using the
Protocol 2: Fine-Tuning a Universal MLIP for Specific Materials
When a pre-trained universal MLIP (e.g., CHGNet, MACE-MP-0) shows inaccuracies for a specific system, fine-tuning on a targeted DFT dataset can restore accuracy [47].
- Identify the Failure Mode:
- Run a test simulation, such as a structural relaxation or
NPTMD simulation of a phase transition (e.g., the PTO-test for PbTiO₃). - Identify the specific inaccuracy, such as incorrect lattice parameters or failure to reproduce a known phase transition [47].
- Run a test simulation, such as a structural relaxation or
- Generate Targeted Training Data:
- Perform DFT calculations (using an appropriate XC functional) to create a dataset that captures the relevant physics. This should include:
- Energy-volume curves around the equilibrium volume.
- Non-equilibrium structures from
NVTMD trajectories at relevant temperatures. - Explicit supercell calculations for key phonon modes or defect configurations.
- Perform DFT calculations (using an appropriate XC functional) to create a dataset that captures the relevant physics. This should include:
- Fine-Tuning Process:
- Start with the pre-trained weights of the universal MLIP.
- Continue training for a limited number of epochs (e.g., 50-100) on the new, targeted dataset, using a low learning rate (e.g., 10⁻⁵).
- Use a 90/10 split for training and validation to monitor for overfitting.
- Validation:
- The fine-tuned model (e.g., MACE-FT) should be validated on properties not explicitly included in the fine-tuning dataset, such as the temperature-driven phase transition pathway or vacancy migration barriers [47].
Protocol 3: Phonon Calculation for a System with a Vacancy
This protocol details the steps for a specific SCLD calculation for a system containing a single vacancy, using a universal MLIP for force evaluations.
- Supercell Generation with a Vacancy:
- Start from a conventionally-sized unit cell of the pristine material.
- Use a tool like the
supercellprogram [21] orASEto generate a sufficiently large supercell (e.g.,4x4x4) to minimize vacancy-vacancy interactions under periodic boundary conditions. - Remove a single atom from a symmetrically unique site to create the vacancy defect. The
supercellprogram can automate the generation of unique vacancy configurations for multi-atom bases [21].
- Structural Relaxation:
- Use a universal MLIP (e.g., Mattersim-v1, CHGNet) to relax the atomic coordinates of the supercell containing the vacancy, keeping the lattice constants fixed or allowing them to relax.
- Convergence Criteria: Set force tolerance to < 0.01 eV/Å and energy tolerance to < 10⁻⁵ eV.
- Force Constant Calculation:
- Using the relaxed defective structure, employ the finite-displacement method as implemented in
phonopy. - The universal MLIP is used as the force calculator for each of the symmetrically unique displaced supercells.
- The MLIP computes the Hellmann-Feynman forces for each configuration, which
phonopyuses to build the force constant matrix.
- Using the relaxed defective structure, employ the finite-displacement method as implemented in
- Post-Processing and Analysis:
- Calculate the phonon density of states (DOS) and phonon band structure for the defective supercell.
- Analyze the resulting spectra for:
- The presence of imaginary frequencies, which would indicate dynamical instability.
- The emergence of localized phonon modes around the vacancy site.
- Changes in the low-frequency spectrum compared to the pristine cell, which can impact thermodynamic properties and ion migration [24].
Case Study: Phonons and Defect Photoemission
A landmark study demonstrated the successful application of universal MLIPs to accelerate the calculation of photoluminescence (PL) spectra for point defects [45]. The calculation of the HR factor, which quantifies electron-phonon coupling, requires the phonon modes of the defect-containing supercell—a major DFT bottleneck. The study benchmarked seven universal MLIPs on a dataset of 791 point defects in 2D materials.
- Method: The structural displacement vector, ΔR, between the ground and excited states was computed with DFT. The phonon modes and frequencies for the supercell were then calculated using the MLIPs instead of DFT.
- Finding: The Mattersim-v1 model achieved exceptional agreement with DFT-derived HR factors and PL lineshapes, enabling speed-ups exceeding an order of magnitude.
- Implication: This establishes that universal MLIPs can reliably capture the lattice dynamics of defective systems, making high-throughput screening of defect optical properties computationally tractable [45].
The Scientist's Toolkit
Table 2: Essential Software and Databases for ML-Accelerated SCLD
| Resource Name | Type | Primary Function | Relevance to SCLD & Vacancies |
|---|---|---|---|
| supercell [21] | Software | Generates supercells with substitutional disorder and vacancies. | Systematically creates all symmetry-inequivalent configurations of vacancies in a supercell. [21] |
| phonopy | Software | Performs phonon calculations using the finite displacement method. | Standard tool for post-processing MLIP-computed forces to obtain phonon band structures and DOS. |
| CHGNet, MACE-MP-0, Mattersim-v1 | Pre-trained Models | Universal MLIPs for energy and force prediction. | Drop-in force calculators for phonopy, enabling fast SCLD for pristine and defective crystals. [46] [45] |
| Materials Project [46] | Database | Repository of DFT-calculated crystal structures and properties. | Source of initial structures for generating training data or for direct screening. |
| MDR Phonon Database [46] [44] | Database | Collection of ab initio phonon calculations. | Crucial for benchmarking the performance of MLIPs on phonon properties. [46] |
Mitigating Convergence Issues in Strongly Disordered Systems
Supercell lattice dynamics (SCLD) provides a powerful first-principles framework for investigating phonon properties in crystalline materials. However, its application to strongly disordered systems, such as those with high concentrations of vacancies or complex cationic arrangements, introduces significant convergence challenges. These challenges primarily stem from the breakdown of the harmonic approximation in the presence of substantial mass disorder and force constant fluctuations, which can lead to unstable phonon modes and non-convergent thermal properties. This Application Note details protocols for mitigating these issues, enabling reliable phonon calculations in disordered systems relevant to advanced material design, including high-entropy alloys and functional ceramics.
Key Concepts and Quantitative Data
Strong disorder fundamentally alters a crystal's vibrational landscape. The following table summarizes the core phenomena and their impact on phonon transport, which SCLD calculations must accurately capture.
Table 1: Fundamental Phenomena in Disordered Systems Affecting Phonon Convergence
| Phenomenon | Description | Impact on Phonon Transport & Convergence |
|---|---|---|
| Mass Disorder | Fluctuations in atomic mass due to a random distribution of different elemental species or vacancies [48]. | Introduces point-defect scattering, suppressing phonon lifetimes and can lead to imaginary frequencies in the phonon spectrum if not properly handled [48]. |
| Force Constant Fluctuations | Variations in local bonding environments and interatomic force constants due to configurational disorder [48]. | Causes strong phonon scattering, reduces group velocities, and is a primary source of convergence instability in the dynamical matrix [48]. |
| Vibration-Induced Polarization | In polar solids, atomic vibrations of cations and anions in opposite directions create dipole-dipole interactions and macroscopic electric fields [23]. | Leads to inaccurate force constants and a failure to reproduce the correct Longitudinal Optical (LO) - Transverse Optical (TO) splitting if not treated with a non-analytic correction [23]. |
The quantitative effects of these phenomena are often studied by comparing ordered and disordered structures, as shown in the table below.
Table 2: Comparative Quantitative Data: Ordered vs. Disordered Systems
| Parameter | 3D MAX Phase (Ordered) | 2D MXene (Dimensional Constriction) | High-Entropy MXene (Dimensional + Cationic Disorder) |
|---|---|---|---|
| Primary Disorder Type | Minimal (Crystalline) | Dimensional Confinement | Configurational + Mass + Dimensional [48] |
| Representative Lattice Thermal Conductivity (LTC) at Room Temperature | High (Excellent thermal transport) [48] | Significantly Reduced [48] | Ultra-Low (Further 40-60% reduction from 2D MXene) [48] |
| Dominant Scattering Mechanism | Umklapp processes | Boundary scattering [48] | Combined mass disorder, strain field, and anharmonic scattering [48] |
Experimental Protocols
Protocol: SCLD Calculation with Non-analytic Correction for Polar Disordered Systems
Application: This protocol is essential for obtaining accurate phonon dispersion and LO-TO splitting in insulating or semiconducting disordered systems, such as high-entropy oxides or nitrides, where polar effects are significant [23].
Workflow Diagram: SCLD Workflow for Polar Disordered Systems
Detailed Methodology:
Supercell Generation & Structural Optimization:
- Construct a supercell commensurate with the desired wavevector grid. For disordered systems, use Special Quasirandom Structures (SQS) to best approximate a random configuration.
- Employ first-principles codes (e.g., VASP [48]) with the Projector Augmented Wave (PAW) method [48] and a generalized gradient approximation (GGA) functional like PBE [48] to fully relax the supercell's atomic positions and lattice parameters. Use a high energy cutoff (e.g., 550 eV [48]) and a k-point mesh fine enough for energy convergence (e.g., 0.03 Å⁻¹ precision [48]).
Finite-Displacement Force Calculations:
- Using the optimized structure, employ the frozen-phonon (small-displacement) approach. Create a set of supercells where each symmetry-inequivalent atom is displaced by a small amount (typically ~0.01 Å) in positive and negative directions along Cartesian axes.
- For each displaced configuration, perform a single-point first-principles calculation to obtain the Hellmann-Feynman forces on every atom in the supercell.
Construct Real-Space Analytic Force Constants (Φ):
- The real-space interatomic force constant matrix
Φis calculated from the force responses. The elementΦ_{αβ}(j,k,P,Q)represents the force in directionαon atomjin primitive cellPwhen atomkin primitive cellQis displaced in directionβ. - This is computed under the condition of a zero macroscopic electric field and constitutes the analytic part of the force [23].
- The real-space interatomic force constant matrix
Compute Non-analytic Correction (C):
- The non-analytic correction accounts for the long-range Coulomb interactions arising from vibration-induced polarizations. It is added to the dynamical matrix in reciprocal space [23].
- This term requires the Born effective charge tensors
Z*for each atom and the high-frequency dielectric tensorε_∞, which must be calculated from separate density functional perturbation theory (DFPT) calculations on the primitive cell. - The correction is direction-dependent and is crucial for accurately reproducing the LO-TO splitting at the Brillouin zone center [23].
Build & Diagonalize Full Dynamical Matrix:
- The full dynamical matrix
D(q)for a wavevectorqis constructed as:D(q) = D_analytic(q) + C(q)whereD_analytic(q)is the Fourier transform of the real-space force constantsΦ, andC(q)is the non-analytic correction. - Diagonalize
D(q)to obtain the square of the phonon frequenciesω²(q)and the corresponding eigenvectors for each wavevector.
- The full dynamical matrix
Phonon DOS & Thermal Property Calculation:
- Interpolate phonon frequencies onto a very dense mesh of q-points in the Brillouin Zone to compute the Phonon Density of States (DOS).
- Use the phonon DOS to calculate thermodynamic properties like Helmholtz free energy, entropy, and heat capacity within the quasi-harmonic approximation. For lattice thermal conductivity, third-order force constants are needed to compute three-phonon scattering rates [23].
Protocol: Configuration Averaging for Convergent Properties in Highly Disordered Systems
Application: This protocol ensures that calculated properties like phonon DOS and thermal conductivity are representative of the true disordered state and are not biased by a single, potentially atypical, supercell configuration.
Workflow Diagram: Configuration Averaging Protocol
Detailed Methodology:
Generate Multiple Independent SQS: Create several different SQS supercells of the same size and composition, each representing a different random instantiation of the disorder. The number of configurations depends on the system's volatility, but 5-10 is a typical starting point.
Perform SCLD for Each SQS: Execute the complete SCLD protocol (Steps 1-6 from Protocol 3.1) for each generated SQS configuration.
Calculate Target Property for Each SQS: From the phonon DOS of each configuration, compute the property of interest (e.g., vibrational free energy, specific heat, or thermal conductivity).
Compute Arithmetic Mean of Properties: The final, converged value for the property is the arithmetic average over all configurations. The standard deviation across configurations provides an estimate of the uncertainty due to configurational sampling.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software Tools for SCLD in Disordered Systems
| Software / Code | Primary Function | Key Features for Disordered Systems |
|---|---|---|
| VASP [48] [23] | First-Principles Electronic Structure | Uses PAW method [48]; provides forces, total energies, and DFPT-calculated Born charges and dielectric constants needed for non-analytic corrections [23]. |
| Phonopy [23] | Phonon Calculations | Implements the direct (frozen-phonon) approach; can post-process force sets from VASP to obtain force constants and phonon spectra; compatible with large supercells for disorder [23]. |
| ShengBTE [23] | Thermal Conductivity Calculation | Solves the Boltzmann Transport Equation for phonons; requires second- and third-order force constants as input to compute lattice thermal conductivity in disordered and anharmonic materials [23]. |
| ALAMODE [23] | Lattice Anharmonicity | Designed to extract harmonic and anharmonic force constants from first-principles, which is critical for accurately capturing thermal properties in strongly disordered and anharmonic systems [23]. |
| YPHON [23] | Phonon Spectrum & LO-TO Splitting | Implements the mixed-space approach [23], which is critical for correctly handling the long-range Coulomb interaction in polar disordered materials. |
Best Practices for Handling Correlated Vacancy Distributions
The study of correlated vacancy distributions is pivotal for advancing our understanding of ionic transport in materials for energy applications, particularly within the framework of supercell lattice dynamics (SCLD). Vacancies, or the absence of atoms in a crystal lattice, are not always isolated defects but can exhibit correlations that significantly influence material properties, including ionic conductivity and diffusion pathways [49] [50]. In superionic conductors, the interaction between vacancies and the host lattice's vibrational properties—its phonons—can dictate the efficiency of ion transport [24]. This document outlines application notes and protocols for investigating these correlated vacancy distributions using SCLD, providing a standardized methodology for researchers in computational materials science and drug development where solid-state properties impact delivery systems.
The dynamics of lattice vibrations provide a powerful lens through which to view vacancy behavior. Research on sodium superionic conductors has established a strong positive correlation between the phonon mean squared displacement (MSD) of Na+ ions and their diffusion coefficients [24]. This implies that the collective vibrational modes of the lattice can promote or hinder vacancy-mediated ion migration. Furthermore, the interaction energy between a vacancy and a solute atom, known as the binding energy, is a critical metric for quantifying correlations [49]. Accurately calculating these energies in complex alloys, such as Si1-xGex, requires sophisticated approaches like the special quasirandom structures (SQS) method to capture the random local environments that influence defect energetics [50]. The protocols herein are designed to integrate these considerations into a robust workflow for SCLD analysis.
Computational Methodology
Foundation of Supercell Lattice Dynamics with Vacancies
Supercell lattice dynamics calculations involve constructing a periodically repeating unit cell that is large enough to host a defect, such as a vacancy, without introducing significant interaction with its periodic images. The core of this methodology lies in computing the interatomic force constants (IFCs), which describe the relationship between atomic displacements and the resulting forces in the lattice. When a vacancy is introduced, it disrupts the local symmetry and alters the IFCs, leading to changes in the phonon spectrum. These changes can be quantified by examining properties such as the phonon density of states (DOS) and the mean squared displacement (MSD) of atoms, which are directly linked to ionic mobility [24].
For studying correlated distributions, one must consider the interaction between multiple vacancies or between a vacancy and other solute atoms. This requires calculating the binding energy of vacancy-solute pairs or vacancy-vacancy pairs. The binding energy ((E{bind})) for a vacancy-solute pair, for instance, can be determined using the following general formula [50]: (E{bind}(V-X) = E(Supercell{with\ V\ and\ X}) + E(Perfect\ Supercell) - E(Supercell{with\ V}) - E(Supercell_{with\ X})) A negative binding energy indicates an attractive interaction, favoring correlation, while a positive value suggests repulsion. In random alloys, this calculation must be repeated over numerous distinct local atomic environments to obtain a statistically significant average, a process greatly accelerated by the SQS approach [50].
Key Quantitative Parameters and Data Presentation
The table below summarizes the key quantitative parameters that should be extracted from SCLD calculations to characterize correlated vacancy distributions.
Table 1: Key Quantitative Parameters for Characterizing Correlated Vacancy Distributions
| Parameter | Description | Computational Method | Significance |
|---|---|---|---|
Binding Energy (E_bind) [49] [50] |
Energy change associated with forming a vacancy-solute or vacancy-vacancy complex. | DFT-based supercell calculations using eq. (1). | Quantifies the thermodynamic driving force for vacancy correlation; negative values indicate attraction. |
| Phonon Mean Squared Displacement (MSD) [24] | Measure of the average square displacement of an atom from its equilibrium position due to thermal vibrations. | Derived from phonon mode analysis or molecular dynamics simulations. | Strongly correlated with ionic diffusion coefficients; a larger MSD suggests higher ionic conductivity. |
Vacancy Formation Energy (E_f) |
Energy required to form an isolated vacancy in the lattice. | DFT calculation of the energy difference between pristine and defected supercell, corrected for the chemical potential of the removed atom. | Determines the equilibrium concentration of vacancies under specific conditions. |
| Acoustic Cutoff Frequency [24] | The highest frequency of the acoustic phonon branches. | Calculated from the phonon dispersion relations. | Lattice softness (low cutoff frequency) promotes superionic conductivity and is a key lattice dynamics signature. |
Table 2: Lattice Dynamics Signatures Linked to Enhanced Ionic Conductivity [24]
| Lattice Dynamics Feature | Correlation with Ionic Transport |
|---|---|
| Low Acoustic Cutoff Phonon Frequencies | Strong positive correlation; indicates a softer lattice that facilitates ion migration. |
| Low Center of Na+ Vibrational Density of States (VDOS) | Promotes large ion displacement; slightly higher than acoustic cutoff frequencies is optimal. |
| Enhanced Low-Frequency Vibrational Coupling | Coupling between mobile ions and host sublattice promotes collective migration mechanisms. |
| Dominant Low-Frequency Acoustic/Optic Modes | Only a small subset of low-frequency modes contribute significantly to large MSDs and ion migration. |
Experimental Protocols
Protocol 1: Binding Energy Calculation for a Vacancy-Solute Pair
This protocol details the steps for calculating the binding energy between a vacancy and a solute atom in a binary system like Al-Sc or SiGe, using density functional theory (DFT).
1. Supercell Generation: * Start with a fully optimized primitive cell of the host material. * Create a supercell of sufficient size (e.g., 3x3x3 for fcc structures, containing 108 atoms) to minimize periodic image interactions [49]. * For random alloys (e.g., Si1-xGex), employ the Special Quasirandom Structure (SQS) method to generate a supercell that best mimics the pair and multi-site correlation functions of a perfectly random alloy [50].
2. Defect Structure Setup:
* Single Solute Supercell (E_X): Place a single solute atom (X) at a substitutional site in the supercell. For SQS, calculate the formation energy for several unique solute sites to account for environmental variance.
* Single Vacancy Supercell (E_V): Remove one host atom from the pristine supercell to create an isolated vacancy.
* Vacancy-Solute Pair Supercell (E_VX): Create a configuration where the vacancy is a nearest neighbor (1NN) or second nearest neighbor (2NN) to the solute atom [49].
3. DFT Calculation and Energy Convergence:
* Relax all atomic positions in the three defect supercells and the pristine supercell (E_perfect) using DFT until the forces on all atoms are below a predefined threshold (e.g., 0.01 eV/Å).
* Ensure consistent computational parameters (pseudopotentials, k-point mesh, energy cutoff) across all calculations. For accurate electronic properties, a hybrid functional like HSE is recommended [50].
* Extract the total energy from each relaxed calculation.
4. Binding Energy Computation:
* Calculate the binding energy using the formula:
E_bind(V-X) = E_VX + E_perfect - E_V - E_X [50].
* A negative result confirms an attractive interaction between the vacancy and the solute.
Protocol 2: Phonon Spectrum and MSD Calculation in a Defected Supercell
This protocol describes how to compute the phonon spectrum and mean squared displacement for a supercell containing a correlated vacancy distribution.
1. Preparation of the Defected Supercell: * Begin with a fully relaxed supercell containing the desired vacancy configuration (e.g., a vacancy pair or a vacancy-solute complex) from Protocol 1.
2. Force Constant Calculation: * Employ the finite displacement method. Create multiple supercells where each atom is displaced by a small amount (e.g., ±0.01 Å) in each Cartesian direction. * Use DFT to calculate the forces on every atom in each displaced configuration. * From the force-displacement relationships, compute the full matrix of interatomic force constants (IFCs). For large supercells, machine-learning potentials (MLPs) like EquiformerV2 can drastically reduce the computational cost while maintaining accuracy [24].
3. Phonon DOS and Dispersion Calculation: * Diagonalize the dynamical matrix, constructed from the IFCs, to obtain the phonon frequencies and eigenvectors across the Brillouin zone. * Plot the phonon dispersion relations and the projected phonon density of states (pDOS), which can reveal the specific contributions of the mobile ions (e.g., Na+) and the host framework.
4. Mean Squared Displacement (MSD) Determination:
* The phonon MSD for atom k can be calculated from the phonon modes using the formula:
MSD_k = (ħ/2N_k) * Σ_i [ (1/ω_i) * |e_i,k|^2 * coth(ħω_i / 2k_B T) ]
where the sum is over all phonon modes i, ωi is the frequency, ei,k is the eigenvector for atom k, and N_k is the number of atoms of type k [24].
* Analyze the correlation between the MSD of the mobile ions and their known diffusion coefficients to validate the model.
Workflow Visualization
The following diagram illustrates the integrated computational workflow for handling correlated vacancy distributions, from supercell preparation to final analysis.
Integrated Computational Workflow for Correlated Vacancy Analysis
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
This section lists the key software and computational "reagents" essential for conducting research on correlated vacancy distributions using SCLD.
Table 3: Key Research Reagent Solutions for SCLD-Vacancy Research
| Tool / Solution | Type | Primary Function | Application Note |
|---|---|---|---|
| VASP [50] | Software Package | First-principles DFT calculations using the projector augmented-wave (PAW) method. | Industry standard for high-accuracy energy and force calculations; essential for defect formation and binding energies. |
| CASTEP [50] | Software Package | Plane-wave DFT code for electronic structure calculations. | Useful for geometry optimization and preliminary phonon calculations; can be coupled with specialized tools. |
| SQS Method [50] | Computational Algorithm | Generates small supercells that mimic the correlation functions of a random alloy. | Critical for studying defects in non-ordered alloys like Si1-xGex; reduces number of configurations needed. |
| EquiformerV2 (MLP) [24] | Machine Learning Potential | A machine-learned interatomic potential trained on DFT data. | Dramatically accelerates force field and lattice dynamics calculations for large systems and long time scales. |
| HSE Hybrid Functional [50] | Computational Method | Exchange-correlation functional mixing Hartree-Fock and DFT (PBE). | Corrects the band gap underestimation of standard DFT, leading to more accurate electronic properties. |
| Phonopy | Software Package | A tool for performing phonon calculations based on the finite displacement method. | Widely used for post-processing force constants to obtain phonon DOS, dispersion, and thermodynamic properties. |
The accurate calculation of phonons—the quantized lattice vibrations in crystalline materials—is fundamental to predicting a wide range of material properties, including thermal conductivity, phase stability, and spectroscopic behavior. For decades, the supercell lattice dynamics (SCLD) approach, particularly through the frozen-phonon (or finite-displacement) method, has been the established computational standard for these calculations. This method operates by creating a supercell of the primitive crystal unit cell, systematically displacing atoms from their equilibrium positions, and using density functional theory (DFT) to calculate the resulting forces. These forces are used to construct the interatomic force constants (IFCs) and ultimately solve for the phonon frequencies and eigenvectors [51] [52]. While this method is considered a benchmark for accuracy, it is computationally intensive, as it requires DFT calculations on numerous supercells, especially for systems with many atoms or low symmetry [52].
Recent advances in computational materials science have spurred the development of alternative methods that seek to reduce this computational burden while maintaining acceptable accuracy. These include compressive sensing lattice dynamics (CSLD), machine learning interatomic potentials (MLIPs), and methods leveraging specific physical insights, such as the minimal molecular displacements (MMD) for molecular crystals [51] [4] [52]. This application note provides a structured framework for researchers to select the most appropriate phonon calculation methodology based on their specific accuracy requirements, computational resources, and material system. We detail explicit protocols, provide quantitative comparisons, and outline decision pathways to guide this critical choice, with special consideration for systems with point defects like vacancies.
Comparative Analysis of SCLD and Alternative Methods
The choice between SCLD and alternative methods involves a direct trade-off between computational cost and predictive accuracy. The following table summarizes the key characteristics, strengths, and limitations of each approach.
Table 1: Comparison of SCLD and Alternative Phonon Calculation Methods
| Method | Computational Cost | Key Strengths | Primary Limitations | Ideal Use Cases |
|---|---|---|---|---|
| SCLD (Frozen-Phonon) | High (Many DFT supercell calculations required) | Considered a gold standard for accuracy; systematically improvable; directly from first principles [52]. | Computationally prohibitive for large/complex systems; cost scales poorly with cell size/symmetry [52]. | Benchmarking; small unit cells; final production calculations for publications. |
| Compressive Sensing (e.g., HiPhive, ALAMODE) | Moderate (Reduced number of DFT calculations via fitting) | Efficiently extracts high-order IFCs; suitable for anharmonic properties like thermal conductivity [4] [53]. | Accuracy depends on training set quality; fitting process can be complex to automate. | High-throughput screening; anharmonic property calculation (κL, CTE) [4]. |
| Machine Learning Interatomic Potentials (MLIPs) | Low (After model training) / High (For training data generation) | Near-DFT accuracy at orders-of-magnitude lower cost after training; enables large-scale MD [52] [54]. | Requires significant training data; risk of poor transferability to unseen configurations. | High-throughput screening of chemically similar compounds; large systems [24] [52]. |
| Minimal Molecular Displacements (MMD) | Low (4-10x reduction vs. SCLD for molecular crystals) [51] | Physical insight reduces computational cost; excellent for low-frequency thermal phonons [51]. | Specialized for molecular crystals; some accuracy loss at high frequencies. | Molecular crystals, organic semiconductors [51]. |
For research on systems with vacancies, the choice of method is critical. SCLD requires a sufficiently large supercell to isolate the defect and prevent spurious interactions between periodic images, which can make the calculation extremely expensive [55]. MLIPs trained on configurations that include defective structures offer a promising path to efficiently model the lattice dynamics around vacancy sites, as they can capture the local force field changes without needing a large supercell for every new calculation [52] [54].
Experimental Protocols
Protocol for Traditional SCLD with Frozen-Phonon Method
This protocol is designed for obtaining high-accuracy harmonic phonon dispersions using the finite-displacement method, serving as a benchmark or for final production calculations.
Table 2: Key Research Reagent Solutions for SCLD
| Item | Function / Recommendation | Technical Notes |
|---|---|---|
| DFT Code | VASP, Quantum ESPRESSO, ABINIT | Must provide accurate forces and energies. |
| Phonopy Package | Primary tool for generating displacements and post-processing force constants [4]. | Industry standard for SCLD. |
| Exchange-Correlation Functional | PBEsol | Recommended over PBE for more accurate lattice parameters and phonon frequencies [4]. |
| Supercell Size | >20 Å cell length or convergence testing | Must be large enough to decay long-range interactions. |
| Atomic Displacement | 0.01 Å – 0.05 Å | Small, finite displacement to remain in harmonic regime. |
Procedure:
- Structure Optimization: Perform a stringent geometry optimization of the primitive cell using a solid-optimized functional like PBEsol until forces on atoms are below 1 meV/Å and stresses are minimized.
- Supercell Construction: Use Phonopy to create a supercell from the optimized primitive cell. The supercell size must be chosen to ensure phonon properties are converged; a minimum dimension of 20 Å is a typical starting point [4].
- Generate Displacements: Using the finite-displacement method within Phonopy, generate a set of supercells where each symmetry-inequivalent atom is displaced in the positive and negative directions of each Cartesian axis.
- DFT Force Calculations: Perform a single-point DFT calculation (no electronic relaxation) for each displaced supercell to compute the Hellmann-Feynman forces on every atom. These calculations are "embarrassingly parallel" and can be run simultaneously.
- Force Constant Calculation: Provide the calculated forces back to Phonopy, which will construct the harmonic force constant matrix.
- Phonon Property Calculation: Use the force constants to calculate phonon band structures, density of states, and thermodynamic properties.
Protocol for High-Throughput Screening with MLIPs
This protocol leverages machine learning to accelerate the screening of phonon properties across many materials, ideal for identifying promising candidates for further study.
Procedure:
- Training Data Generation:
- Select a diverse set of representative structures from your target chemical space.
- For each structure, generate 4-8 supercells with all atoms randomly perturbed by displacements between 0.01 Å and 0.05 Å from their equilibrium positions [52].
- Perform DFT calculations on these perturbed supercells to obtain the total energy and interatomic forces for each configuration.
- Model Training:
- Choose a state-of-the-art MLIP architecture, such as MACE (Message Passing Neural Network with Atom-Centered Symmetrized Features) [52].
- Train the model on the dataset of perturbed structures, energies, and forces. The model learns the underlying potential energy surface (PES).
- Phonon Prediction:
- For a new, unseen crystal structure, use the trained MLIP to predict forces for a set of small atomic displacements, bypassing the need for explicit DFT calculations.
- Feed these predicted forces into a lattice dynamics code (e.g., Phonopy) to compute the phonon spectrum.
This workflow can achieve a significant speed-up while maintaining high accuracy, with one study reporting a mean absolute error of less than 40 cm⁻¹ for phonon frequencies across a diverse dataset [52].
Decision Framework and Workflow Visualization
Selecting the optimal method requires evaluating the research objective, material system, and available resources. The following decision pathway provides a visual guide for this process.
Diagram 1: Phonon Method Decision Pathway
The workflow for calculating phonons, once a method is selected, follows a general pattern of moving from initial structure preparation to the final computation of properties. The SCLD and CSLD methods share a common high-level structure but differ significantly in the intermediate steps, as visualized below.
Diagram 2: SCLD vs. CSLD Workflow Comparison
The landscape of computational lattice dynamics offers multiple paths forward. The traditional SCLD approach remains the method of choice for benchmark-quality results and systems where computational cost is secondary to accuracy. However, for high-throughput discovery, studies of complex systems like those with vacancies, or investigations of strongly anharmonic materials, modern alternatives like CSLD and MLIPs are no longer just approximations but powerful and often necessary tools. By applying the decision framework and protocols outlined in this document, researchers can strategically balance accuracy and efficiency to accelerate their research without compromising scientific rigor.
Benchmarking and Validation: Ensuring Accuracy in SCLD Predictions for Vacancy Systems
In computational materials science, accurately predicting the vibrational properties (phonons) of defective crystals, such as those with vacancies, is crucial for understanding thermal conductivity, phase stability, and mechanical behavior. Two predominant methodologies for these calculations are Supercell Lattice Dynamics (SCLD) and the Virtual Crystal Approximation (VCA). SCLD models defects explicitly within a large, repeated supercell, directly calculating their impact on the lattice dynamics. In contrast, VCA employs a mean-field approach, creating a hypothetical average crystal to represent disordered systems or defects, offering computational efficiency but potentially at the cost of physical accuracy.
This application note provides a detailed comparison of these two approaches, framed within research on phonon calculations in systems with vacancies. We include structured protocols, data comparisons, and decision frameworks to guide researchers in selecting and applying the appropriate method.
Supercell Lattice Dynamics (SCLD)
SCLD is a direct approach for modeling defects in crystalline materials. A supercell—a large repetition of the primitive crystal unit cell—is constructed, and vacancies or other defects are introduced explicitly at specific atomic sites. The phonon properties are then calculated by solving the equations of motion for this large, defective system. This method can accurately capture the local structural distortions and force-constant changes induced by the defect.
Virtual Crystal Approximation (VCA)
VCA is a mean-field technique that models a disordered alloy or a defective crystal as an ordered structure with "virtual" atoms. The potential of these virtual atoms is constructed as a compositionally weighted average of the constituent elements or, in the case of vacancies, a perturbation from the perfect crystal potential. For example, in a system with carbon vacancies, the virtual atom potential ( V{VCA} ) might be expressed as ( (1-c)V{C} + c V_{Vacancy} ), where ( c ) is the vacancy concentration [56]. This method is technically simpler and computationally less expensive, as it retains the cost of a calculation for a primitive unit cell.
Comparative Analysis: SCLD vs. VCA
The choice between SCLD and VCA involves a fundamental trade-off between computational cost and predictive accuracy. The table below summarizes their core characteristics based on a study of solid-solution refractory metal carbides with carbon vacancies [56].
Table 1: A comparative overview of SCLD and VCA methodologies.
| Feature | Supercell Lattice Dynamics (SCLD) | Virtual Crystal Approximation (VCA) |
|---|---|---|
| Fundamental Approach | Direct, explicit inclusion of defects | Mean-field approximation of an average crystal |
| Model System | Large supercell with specific atomic vacancies | Primitive cell with pseudo-atoms representing an average potential |
| Computational Cost | High (scales with supercell size) | Low (equivalent to a primitive cell calculation) |
| Treatment of Vacancies | Atomistic and local | Smoothed and delocalized |
| Accuracy for Localized Phenomena | High, captures local symmetry breaking | Low, can fail to describe properties reliant on local atomic environments |
| Key Limitation | Computationally prohibitive for very low defect concentrations | Underestimates lattice parameter and overestimates elastic constants in defective carbides [56] |
Quantitative findings from a study on refractory metal carbides highlight these differences. The research employed a "similar atomic environment" supercell method to benchmark VCA's performance [56]:
Table 2: Quantitative comparison of VCA and supercell method predictions for selected solid-solution carbides [56].
| Solid-Solution Carbide | Property | VCA Prediction | Supercell (SAE) Prediction |
|---|---|---|---|
| (Ti~0.5~Zr~0.5~)C | Lattice Parameter (Å) | Underestimated | Accurate to experimental trends |
| (Ti~0.5~Nb~0.5~)C | Elastic Constants | Overestimated | Softer, more accurate values |
| (V~0.5~Nb~0.5~)C | Mechanical Failure Mode | Incorrect for vacancies | Captures weakening effect of vacancies |
The study concluded that while VCA is a valuable tool for initial, high-throughput screening, SCLD (or similar supercell approaches) is necessary for obtaining quantitatively accurate mechanical and dynamical properties in systems with point defects like vacancies [56].
Experimental and Computational Protocols
Protocol for SCLD Phonon Calculation with Vacancies
This protocol details the steps for performing a phonon calculation for a system with vacancies using the SCLD method.
4.1.1 Research Reagent Solutions
Table 3: Essential software and computational tools for SCLD calculations.
| Item Name | Function & Application |
|---|---|
| DFT Code (VASP, Quantum ESPRESSO) | Performs first-principles electronic structure calculations to determine the total energy and Hellmann-Feynman forces of the supercell. |
| Phonopy Software | Post-processes the force constants obtained from DFT calculations to compute phonon dispersion spectra and density of states. |
| Supercell Construction Tool | Used to build the defective supercell from the primitive cell. This can be done with codes like ASE or built-in tools in materials studio. |
4.1.2 Step-by-Step Workflow
Supercell Generation:
- Start with a fully relaxed primitive unit cell of the host material.
- Construct a supercell of sufficient size (e.g., 3x3x2 or 4x4x4) to minimize the interaction between periodic images of the vacancy. The supercell should contain N atoms.
- Introduce a vacancy by removing a single atom from a chosen lattice site, resulting in a system with N-1 atoms.
Geometry Optimization:
- Use Density Functional Theory (DFT) to relax the ionic positions of the defective supercell, allowing atoms around the vacancy to find their new equilibrium positions. The lattice vectors are typically held fixed.
Force Calculation:
- In the optimized structure, displace each atom in the supercell by a small finite amount (e.g., ±0.01 Å) in the positive and negative directions of each Cartesian axis.
- For each displacement, perform a single-point DFT calculation to record the forces on every other atom in the supercell. This generates a force constant matrix.
Phonon Post-Processing:
- Use the set of calculated forces as input for phonon software like Phonopy.
- The software constructs the dynamical matrix and diagonalizes it to obtain the phonon frequencies and eigenvectors across the Brillouin zone.
Diagram 1: SCLD phonon calculation workflow for a system with a vacancy.
Protocol for VCA Phonon Calculation
This protocol outlines the steps for a phonon calculation using the VCA approach, which is computationally less intensive.
4.2.1 Step-by-Step Workflow
Virtual Crystal Construction:
- Define a "virtual" atom species for the defective crystal. For a system with a carbon vacancy concentration
c, the pseudopotential for the virtual carbon site ( V{VCA} ) is generated as a linear combination: ( V{VCA} = (1-c)V{C} + c V{Vacancy} ) [56]. The vacancy potential ( V_{Vacancy} ) is often approximated or set to zero.
- Define a "virtual" atom species for the defective crystal. For a system with a carbon vacancy concentration
Cell Relaxation:
- Use DFT with the newly generated VCA pseudopotential to relax the lattice parameter and ionic positions of the primitive unit cell. This yields an averaged equilibrium structure.
Phonon Calculation:
- Perform a standard phonon calculation on the relaxed VCA primitive cell using Density Functional Perturbation Theory (DFPT) or the finite displacement method. Due to the periodicity and lack of explicit defects, this is computationally efficient.
Diagram 2: VCA phonon calculation workflow for a defective system.
The decision to use SCLD or VCA hinges on the specific research goal, the nature of the defect, and available computational resources.
Diagram 3: Decision framework for selecting between SCLD and VCA.
5.1 When to Use SCLD:
- High-Accuracy Requirements: When quantitative accuracy for elastic constants, phonon densities of states, and mechanical properties is critical [56].
- Localized Phenomena: To investigate the localized vibrational modes introduced by the vacancy or the breakdown of translational symmetry.
- Low/Medium Defect Concentrations: Where the supercell can be made large enough to make periodic defect-defect interactions negligible.
5.2 When to Use VCA:
- High-Throughput Screening: For rapid preliminary studies across a wide range of compositions or vacancy concentrations.
- Trend Analysis: To identify general trends in properties like lattice parameter or bulk modulus with changing defect concentration.
- Computational Resource Constraints: When system size makes SCLD calculations prohibitively expensive.
In conclusion, SCLD remains the gold standard for accuracy in phonon calculations with vacancies, directly capturing the localized physics of the defect. VCA offers a computationally efficient alternative for studying average properties and trends but suffers from inaccuracies in predicting key mechanical and dynamical properties. A combined approach, using VCA for initial screening and SCLD for detailed analysis of promising systems, often represents the most effective research strategy.
This document provides detailed application notes and protocols for two pivotal spectroscopic techniques—Inelastic Neutron Scattering (INS) and Raman Spectroscopy. These techniques are essential for experimental validation within research frameworks focusing on supercell lattice dynamics (SCLD) for phonon calculations in materials containing vacancies. Both methods probe vibrational excitations but operate on different physical principles and selection rules, making them highly complementary [57] [58]. For researchers investigating defect physics, the combination of INS and Raman spectroscopy offers a comprehensive picture of the perturbed phonon density of states and the localized vibrational modes induced by vacancies.
Raman spectroscopy measures the inelastic scattering of monochromatic light, usually from a laser. The interaction of light with molecular vibrations or phonons results in a shift in the energy of the laser photons, providing a structural fingerprint for material identification [57] [59]. The Raman effect relies on a change in the electric dipole-electric dipole polarizability of a material during a vibration [57].
Inelastic Neutron Scattering (INS), in contrast, probes vibrations through the energy transfer that occurs when a beam of neutrons interacts with a sample. The scattering cross-section for neutrons is directly related to the nucleus and the vibrational amplitude, allowing for the determination of the generalized phonon density of states [60]. A key difference lies in their selection rules; while Raman intensity depends on the polarizability change, INS does not obey the same selection rules, enabling it to detect vibrational modes that may be silent to both IR and Raman spectroscopy [57].
Table 1: Fundamental Comparison of Raman Spectroscopy and Inelastic Neutron Scattering
| Feature | Raman Spectroscopy | Inelastic Neutron Scattering |
|---|---|---|
| Probe Particle | Photon (Laser light) | Neutron |
| Measured Quantity | Energy shift of scattered photon (Raman shift) | Energy transfer of scattered neutron |
| Key Selection Rule | Change in polarizability | No optical selection rules |
| Primary Output | Raman spectrum (Intensity vs. Wavenumber) | Scattering function S(q,ω), Generalized Density of States (GDOS) |
| Sensitivity to Vacancies | Detection of local vibrational modes and symmetry changes | Direct measurement of changes in full phonon density of states |
Table 2: Typical Experimental Parameters for SCLD Validation Studies
| Parameter | Raman Spectroscopy | Inelastic Neutron Scattering |
|---|---|---|
| Typical Excitation Source | 532 nm laser (visible) [59] | Incident neutron wavelength, e.g., λ = 4 Å [60] |
| Sample Environment | Ambient conditions, various temperatures | Often requires cryogenic temperatures (e.g., 4 K) to high T (e.g., 1000 K) [60] |
| Data Collection Time | Seconds to minutes | Hours to days |
| Key Data Correction Steps | Filtering of Rayleigh scattering [57] | Normalization to vanadium standard, empty scatter correction [60] |
| Phonon Information | Zone-center (Γ-point) optical phonons | Full Brillouin zone phonon dispersion |
Experimental Protocols
Protocol for Raman Spectroscopy
3.1.1 Principle When light interacts with a substance, most photons are elastically scattered (Rayleigh scattering). A tiny fraction (~1 in 10 million) undergoes inelastic scattering, where the photon loses (Stokes shift) or gains (anti-Stokes shift) energy equal to the vibrational energy of a molecular bond or phonon [59]. This energy shift is independent of the excitation laser wavelength and provides a unique vibrational fingerprint [58].
3.1.2 Sample Preparation
- Solid Samples: Samples can be analyzed as single crystals, polycrystalline powders, or thin films. Little to no preparation is often needed [59].
- Containment: A key advantage of Raman spectroscopy is the ability to analyze samples through transparent packaging like glass vials or plastic bags, as visible laser light can penetrate these materials [59].
- Considerations: The sample should ideally be non-fluorescent. For sensitive materials, laser power must be minimized to avoid damage through heating or photodecomposition [58] [59].
3.1.3 Instrumentation and Data Acquisition
- Laser Selection: Illuminate the sample with a monochromatic laser source. Common wavelengths include 532 nm (green) or 785 nm (NIR). Near-infrared lasers help mitigate fluorescence [57] [59].
- Light Collection: Collect the scattered light from the sample with a lens.
- Rayleigh Rejection: Filter out the intense, elastically scattered Rayleigh light using a notch filter or edge pass filter [57].
- Dispersion and Detection: Disperse the remaining inelastically scattered light onto a charge-coupled device (CCD) detector to record the spectrum [57].
3.1.4 Data Processing
- Pre-processing: Apply instrument-specific calibration and may include cosmic ray removal and baseline correction.
- Peak Analysis: Identify the Raman shifts (in cm⁻¹) of all observed peaks. The shift is calculated as Δν̃ = (1/λ₀ - 1/λ₁) × 10⁷, where λ₀ and λ₁ are the excitation and scattered wavelengths in nm, respectively [57].
- Interpretation: Compare the spectrum to reference spectral libraries. Peaks are assigned to specific vibrational modes, symmetry changes, or localized modes induced by vacancies [59].
Protocol for Inelastic Neutron Scattering
3.2.1 Principle A beam of monoenergetic neutrons is directed at the sample. Neutrons interact with atomic nuclei, and upon scattering, they gain or lose energy and momentum by creating or annihilating phonons. This process provides a direct measure of the phonon density of states [60].
3.2.2 Sample Preparation
- Large Masses: Due to relatively weak neutron scattering cross-sections, gram-scale quantities of sample are typically required.
- Deuterated Containers: Where possible, use deuterated sample containers or environments to minimize the overwhelming parasitic scattering from hydrogen atoms.
- Characterization: The sample should be well-characterized (e.g., phase purity, vacancy concentration) prior to the INS experiment.
3.2.3 Instrumentation and Data Acquisition This protocol is based on a typical experiment performed on a time-of-flight spectrometer like FOCUS [60].
- Sample Loading: Load the pristine and vacancy-containing samples into the spectrometer.
- Experimental Conditions: Conduct measurements at relevant temperatures (e.g., 400 K, 800 K, 1000 K) to probe temperature-dependent phonon effects [60].
- Neutron Scattering: Expose the sample to an incident neutron beam with a defined wavelength (e.g., λ = 4 Å). Measure the energy transfer up to a specified maximum (e.g., 100 meV).
- Calibration: Collect and subsequently subtract data for an empty container to correct for background scattering.
- Normalization: Normalize all measured spectra to the scattering from a standard vanadium sample [60].
3.2.4 Data Processing
- Data Reduction: Use specialized software (e.g., the DAVE package) to convert the raw time-of-flight data into the scattering function S(q,ω) [60].
- GDOS Extraction: Derive the generalized phonon density of states (GDOS) from S(q,ω) [60].
- Comparative Analysis: Directly compare the GDOS of the pristine sample with that of the sample containing vacancies. Differences reveal the impact of vacancies on the overall lattice dynamics.
Workflow Visualization
Figure 1: Integrated experimental workflow for SCLD model validation.
Figure 2: Raman spectroscopy measurement and data flow.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials and Reagents for INS and Raman Studies
| Item | Function/Description | Key Consideration |
|---|---|---|
| Monochromatic Laser | Provides the excitation source for Raman spectroscopy. Common wavelengths: 532 nm, 785 nm, 1064 nm. | Shorter wavelengths yield stronger scattering but may cause fluorescence/sample damage. NIR (1064 nm) mitigates fluorescence [59]. |
| Notch/Edge Filter | Optical filter that removes the intense elastically scattered (Rayleigh) laser light. | Critical for detecting the weak Raman signal which is very close in wavelength to the laser line [57]. |
| CCD Detector | The standard detector for dispersive Raman spectrometers, used to record the spectrum. | High sensitivity and low noise are required due to the inherent weakness of the Raman effect [57]. |
| Time-of-Flight Neutron Spectrometer | Instrument used for INS experiments; measures neutron energy by time of flight. | Provides access to the full phonon spectrum, including acoustic modes [60]. |
| Vanadium Standard | A calibration sample used for neutron scattering experiments. | Vanadium has a largely incoherent and energy-independent scattering cross-section, making it ideal for normalization [60]. |
| Deuterated Sample Cells | Containers for holding samples during neutron scattering experiments. | Deuterium has a much lower neutron scattering cross-section than hydrogen, reducing background signal. |
| Cryostat/Furnace | Sample environment control for temperature-dependent measurements. | Essential for studying phonon behavior and stability across a wide temperature range (e.g., 400-1000 K) [60]. |
The study of thermal transport in materials with atomic-scale defects, such as vacancies, is crucial for the development of advanced thermoelectrics, electronics, and other functional materials. Within this research domain, two computational methodologies are particularly relevant: Molecular Dynamics (MD) and the emerging Sequential Controlled Langevin Diffusions (SCLD). MD simulations, including non-equilibrium MD (NEMD), provide a direct approach to simulate atomic trajectories and calculate thermal conductivity from statistical mechanics. In contrast, SCLD is a novel sampling-based method that combines ideas from Sequential Monte Carlo and diffusion models to efficiently explore complex energy landscapes. This application note details protocols for employing these methods specifically for investigating vacancy-mediated thermal transport, framing them within a verification paradigm to leverage their complementary strengths.
Core Principles
- Molecular Dynamics (MD): MD simulates the real-time evolution of a system of atoms based on classical mechanics. For thermal transport, a common technique is the Non-Equilibrium Molecular Dynamics (NEMD) approach. This method typically imposes a temperature gradient across the simulation cell using thermostats (e.g., Langevin thermostats) and calculates the resulting heat flux to determine thermal conductivity via Fourier's law [36]. The interatomic interactions are described by empirical potentials or machine-learning-driven force fields.
- Sequential Controlled Langevin Diffusions (SCLD): SCLD is a sampling algorithm designed to generate configurations from a target probability distribution, such as the Boltzmann distribution. It is not a dynamics method in the same sense as MD. Instead, it progressively transports samples from a simple prior distribution to the complex target distribution using a learned stochastic differential equation (SDE). A key feature is its use of resampling steps, which focus computational effort on promising regions of the configuration space, and its ability to be trained end-to-end [61]. Its primary application in this context is to efficiently generate thermodynamically representative atomic configurations, including those with vacancies, from which properties can be computed.
Comparative Analysis
The table below summarizes the key characteristics of both methods for thermal transport studies.
Table 1: Comparison of MD and SCLD for Thermal Transport Analysis
| Feature | Molecular Dynamics (MD) | Sequential Controlled Langevin Diffusions (SCLD) |
|---|---|---|
| Fundamental Approach | Direct simulation of atomic trajectories via numerical integration of equations of motion. | Learned, gradual transport of samples to a target equilibrium distribution via an SDE. |
| Primary Output for Thermal Properties | Thermal conductivity from heat flux/temperature gradient (NEMD) or equilibrium fluctuations (Green-Kubo). | Representative sample configurations; thermal properties require subsequent analysis. |
| Treatment of Vacancies | Explicitly included in the initial atomic model; their effects emerge from the simulated dynamics [62] [63]. | Configurations with vacancies are generated as samples from the target distribution. |
| Key Strength | Direct access to dynamical information and phonon spectra. Intuitive connection to real-time physics. | Potentially more efficient exploration of configuration space and free energy landscapes. Resampling avoids getting trapped in local minima. |
| Computational Cost/ Training | No training required, but can be limited by the time scales accessible by simulation. | Requires an initial training phase to learn the SDE drift, but can then generate samples efficiently [61]. |
| Role in Cross-Verification | Provides a benchmark with established, physics-direct methodology. | Offers an alternative, potentially efficient route to equilibrium properties and can validate MD sampling. |
Experimental and Computational Protocols
Protocol for NEMD Simulation of Defective Structures
This protocol outlines the steps for calculating thermal conductivity in a structure with vacancy defects using NEMD, as employed in studies of twisted multilayer graphene [36].
System Setup:
- Model Construction: Create an atomic model of the system. For a vacancy study, remove one or more atoms from a perfect crystal lattice. For 2D materials like graphene, define the in-plane dimensions and the number of layers [36].
- Force Field Selection: Choose an appropriate interatomic potential. For complex systems, machine-learning potentials like the neuroevolution potential (NEP) are increasingly used to accurately capture interactions, including van der Waals forces in layered materials [36].
- Boundary Conditions: Apply periodic boundary conditions in the in-plane directions (x, y). Use fixed boundary conditions in the cross-plane direction (z) to allow for a temperature gradient [36].
Equilibration:
- Energy minimize the structure to relieve any high-strain configurations introduced by the defects.
- Run an MD simulation in the NVT ensemble (constant Number of particles, Volume, and Temperature) for a sufficient time to equilibrate the system at the desired average temperature (e.g., 300 K).
Production Run (NEMD):
- Thermostat Application: Define two regions within the simulation cell along the direction of heat flow (e.g., the z-axis). Apply a "hot" Langevin thermostat (Th) to one region and a "cold" thermostat (Tc) to another [36].
- Heat Flux Measurement: Run the simulation in the NVE ensemble (constant Number, Volume, and Energy) for the production phase. The heat flux J is equal to the energy added/removed by the thermostats per unit time and area.
- Temperature Gradient: Measure the steady-state temperature profile along the cell. The thermal conductivity κ is calculated using Fourier's law: κ = -J / (dT/dz), where dT/dz is the measured temperature gradient.
Protocol for SCLD-Assisted Sampling
This protocol describes how to use SCLD for sampling configurations of a system with vacancy defects [61].
Problem Formulation:
- Define the target distribution, typically the Boltzmann distribution
p_target ∝ exp(-E(x)/k_B T), whereE(x)is the potential energy of configurationx. - Select a simple prior distribution (e.g., a Gaussian) from which initial samples are drawn.
- Define the target distribution, typically the Boltzmann distribution
Training Phase:
- Parameterization: Parameterize the drift term of the underlying SDE with a neural network.
- Optimization: Train the model using a suitable loss function, such as the log-variance loss, which enables end-to-end training and helps to avoid issues like mode collapse [61]. This phase involves optimizing the neural network to learn the dynamics that transport prior samples to the target distribution.
Inference and Sampling:
- Particle Evolution: Draw a population of "particles" (independent samples) from the prior distribution.
- Sequential Transport & Resampling: Evolve the particles according to the learned SDE. At discrete intervals, perform resampling steps based on importance weights, which helps to focus computational resources on promising trajectories [61].
- Configuration Generation: The final set of particles after the full sequence represents samples from the target distribution. These configurations, which include the thermodynamic effects of vacancies, can be analyzed to compute average properties.
The Scientist's Toolkit: Key Research Reagents and Solutions
Table 2: Essential Computational Tools for Thermal Transport Simulations
| Tool Name | Type | Primary Function | Relevance to Method |
|---|---|---|---|
| GPUMD | Software Package | High-performance MD simulations optimized for GPUs. | MD, NEMD |
| LAMMPS | Software Package | A widely used classical MD simulator with a vast library of force fields. | MD, NEMD |
| Neuroevolution Potential (NEP) | Machine-Learning Potential | Provides accurate forces and energies for MD, including complex interactions. | MD, NEMD [36] |
| SCLD Code (Reference Implementation) | Algorithm/Script | Implements the Sequential Controlled Langevin Diffusion sampling procedure. | SCLD [61] |
| Stochastic Differential Equation (SDE) Solver | Numerical Library | Solves SDEs (e.g., Euler-Maruyama method) for propagating samples. | SCLD [61] |
| Interatomic Force Constants (IFCs) | Data Structure | Describes the harmonic and anharmonic interactions between atoms for lattice dynamics. | Can be output from SCLD/MD for further analysis [4] |
Workflow Visualization
The following diagram illustrates the conceptual workflow for using SCLD and MD in a cross-verification study, highlighting the complementary information each method provides.
Diagram Title: Cross-Verification Workflow for SCLD and MD
Discussion and Outlook
The integration of SCLD and MD presents a powerful framework for advancing the study of thermal transport in defective materials. MD remains the workhorse for direct simulation, providing dynamical information and serving as a benchmark. However, SCLD offers a promising alternative for efficiently sampling the equilibrium configuration space, which is particularly valuable for systems where MD sampling might be slow due to energy barriers or complex defect landscapes [61].
The cross-verification of these methods strengthens the reliability of computational predictions. For instance, the thermal conductivity of a vacancy-containing system calculated via NEMD can be compared to the value derived from the ensemble of configurations generated by SCLD (using methods like lattice dynamics on the sampled structures). Furthermore, SCLD's ability to generate a diverse set of configurations can provide deeper insights into the specific atomic arrangements that most significantly impact phonon scattering and localization, a phenomenon critically important in materials like disordered twisted multilayer graphene [36].
Future work will focus on tighter integration of these methods, such as using representative configurations from SCLD as starting points for MD simulations, or using MD data to inform the training of SCLD models. This synergistic approach, supported by robust protocols as outlined herein, will accelerate the discovery and design of materials with tailored thermal properties.
Application Note: Integrating Machine Learning into SCLD Workflows for Property Prediction
This document provides detailed application notes and protocols for assessing the predictive power of computational methods for two critical material properties—thermal conductivity and thermodynamic stability—within the research context of Supercell Lattice Dynamics (SCLD) for phonon calculations with vacancies.
The Role of Lattice Dynamics and Machine Learning
The foundational principle of this approach is the strong correlation between lattice vibrational properties (phonons) and macroscopic material properties. For instance, in the study of sodium superionic conductors, key lattice dynamics signatures such as low acoustic cutoff phonon frequencies and enhanced low-frequency vibrational coupling have been identified as promoters of superionic conductivity [24]. Quantitative correlations have been established between phonon-derived descriptors, like the phonon mean squared displacement (MSD) of Na+ ions, and ionic diffusion coefficients, providing a physical basis for predictive models [24].
Machine Learning Interatomic Potentials (MLIPs) have emerged as a transformative tool, bridging the gap between the accuracy of quantum-mechanical methods like Density Functional Theory (DFT) and the computational efficiency required for high-throughput screening. These potentials enable the calculation of harmonic phonon properties, which are critical for understanding vibrational and thermal behavior, directly from the second derivatives of the potential energy surface [46].
A Unified Framework for Discovering High Thermal Conductivity Materials
A promising, unified generative deep learning framework has been demonstrated for predicting materials with ultrahigh lattice thermal conductivity (κL) while ensuring stability [64]. This framework integrates several advanced techniques:
- Generative Models: An SE(3)-equivariant Crystal Diffusion Variational Autoencoder (CDVAE) is used for the fast exploration of material space, generating novel, valid crystal structures [64].
- Data Distillation and Active Learning: Generated structures undergo geometry optimization using MLIPs. An active-learning-based protocol, such as Query by Committee (QBC), is employed to selectively improve the MLIP on-the-fly when prediction uncertainty is high, ensuring high-fidelity predictions of stability and κL without the cost of curating massive training datasets [64].
- Efficient Screening: Structural symmetry metrics and similarity analysis are used to filter the vast generated chemical space efficiently. For example, selecting materials with simple unit cells (number of atoms N ≤ 12) and high symmetry (symmetry operations SO ≥ 4) can preferentially retain candidates with high thermal conductivity [64].
When applied to carbon allotropes, this framework successfully identified 34 polymorphs with κL above 800 W m⁻¹ K⁻¹, including previously unpredicted structures, showcasing its predictive power [64].
Experimental Protocols
Protocol 1: High-Throughput Screening of Ionic Conductors via Lattice Dynamics
This protocol outlines the steps for correlating lattice dynamics signatures with ionic transport properties, as demonstrated for sodium superionic conductors [24].
Workflow Overview:
Detailed Methodology:
Initial Structure Curation:
- Source: Extract sodium-containing inorganic crystal structures from the Open Quantum Materials Database (OQMD) and the Inorganic Crystal Structure Database (ICSD) [24].
- Filtering Criteria: Apply filters for a non-zero electronic band gap and a negative formation energy to ensure thermodynamic plausibility [24].
Structural Re-optimization:
- Method: Use Density Functional Theory (DFT) calculations with strict computational parameters to re-optimize the geometry of all filtered structures. This step ensures physically meaningful structural parameters and stable ground-state configurations [24].
Dynamic Stability Screening:
Lattice Dynamics and Molecular Dynamics (MD) Simulations:
- Lattice Dynamics: Using the same MLIP, calculate key lattice dynamics features for the stable structures. These include:
- Phonon Mean Squared Displacement (MSD) of the mobile ions (e.g., Na⁺).
- Acoustic cutoff frequency.
- Center of the vibrational density of states (VDOS) for the mobile ions.
- Low-frequency phonon coupling strength between mobile ions and the host lattice [24].
- MD Simulations: Perform MD simulations using the MLIP to determine the ionic diffusion coefficients of the mobile species [24].
- Lattice Dynamics: Using the same MLIP, calculate key lattice dynamics features for the stable structures. These include:
Correlation and Model Building:
- Analysis: Establish a quantitative correlation between the calculated lattice dynamics descriptors (especially phonon MSD) and the diffusion coefficients obtained from MD [24].
- Machine Learning: Integrate these phonon-derived descriptors into a machine learning framework (e.g., regression models) to rapidly predict ionic transport properties across a broad structural and chemical space [24].
Protocol 2: Active Learning for Thermal Conductivity and Stability Prediction
This protocol describes an active learning loop for the accurate prediction of lattice thermal conductivity (κL) and stability of generated crystal structures, particularly those containing vacancies or defects [64].
Workflow Overview:
Detailed Methodology:
Candidate Generation via Generative Model:
- Tool: Use a generative model like the SE(3)-equivariant Crystal Diffusion Variational Autoencoder (CDVAE) to produce a large number of initial candidate structures. This model learns the joint probability distribution of real materials and can sample new compositions and structures from this distribution [64].
Data Distillation and Initial Screening:
- Structure Optimization: Employ a pre-trained, general-purpose MLIP to perform geometry optimization on all generated structures, ensuring they reside at a local energy minimum [64].
- Deduplication: Conduct structural similarity analysis to remove duplicate structures, retaining only unique candidates [64].
- Symmetry Filtering: Apply a structural symmetry filter to narrow the search space. Empirical rules suggest that simple, high-symmetry unit cells often lead to high thermal conductivity. A practical filter is to retain materials where the number of atoms in the unit cell (N) is ≤ 12 and the number of symmetry operations (SO) is ≥ 4 [64].
Multistep Identification with Active Learning:
- Diverse Benchmark Selection: From the filtered pool, use the Farthest Point Sampling (FPS) algorithm to select a smaller set (m) of structurally diverse benchmarks [64].
- Active Learning ETC Protocol: Implement an active learning protocol for the accurate evaluation of thermal conductivity (ETC) on these benchmarks.
- Use an MLIP committee (multiple models) to predict κL.
- Apply the "Query by Committee" (QBC) strategy: if the prediction uncertainty (e.g., standard deviation between committee members) for a structure exceeds a predefined threshold, that structure is selected for accurate DFT calculation, and the resulting data is used to update and improve the MLIP training set [64].
- This loop continues until model uncertainties are sufficiently low, ensuring high-fidelity κL predictions for the benchmark set.
- Cluster-Based Expansion: Use the k-nearest neighbors (KNN) algorithm to cluster all filtered materials based on structural similarity to the benchmarks. Screen all materials within clusters that contained a high-κL benchmark (e.g., κL ≥ 800 W m⁻¹ K⁻¹) using the now-robust MLIP [64].
Final Validation:
- The final list of predicted ultrahigh κL materials should be validated through direct quantum-mechanical calculations (e.g., DFT with phonon Boltzmann transport theory) or experimental synthesis where feasible [64].
Table 1: Benchmarking Universal MLIPs for Phonon and Stability Predictions [46]
| Model Name | Energy MAE (eV/atom) | Force MAE (eV/Å) | Volume per Atom MAE (ų/atom) | Geometry Optimization Failure Rate (%) | Key Strengths/Weaknesses for SCLD |
|---|---|---|---|---|---|
| CHGNet | ~0.05 (uncorrected) | ~0.05 | ~0.2 | 0.09% | High reliability, smaller architecture, but higher energy error. |
| MatterSim-v1 | Data Not Provided | Data Not Provided | Data Not Provided | 0.10% | Built on M3GNet, uses active learning for broader accuracy. |
| M3GNet | ~0.035 | Data Not Provided | ~0.15 | ~0.2% | A pioneering model, balanced performance. |
| MACE-MP-0 | Data Not Provided | Data Not Provided | Data Not Provided | ~0.2% | Uses atomic cluster expansion for efficiency. |
| SevenNet-0 | Data Not Provided | Data Not Provided | Data Not Provided | ~0.2% | Data-efficient and equivariant, based on NequIP. |
| ORB | Data Not Provided | Data Not Provided | Data Not Provided | 0.67% | Predicts forces separately; may struggle with force convergence. |
| eqV2-M | Data Not Provided | Data Not Provided | Data Not Provided | 0.85% | High accuracy on leaderboards, but highest failure rate in this test. |
Table 2: Thermal Conductivity Enhancement in Composite Phase Change Materials [65]
| Composite PCM System | Graphite Additive & Concentration | Thermal Conductivity (W/m·K) | Enhancement Factor (vs. Pure PCM) | Key Application Finding |
|---|---|---|---|---|
| Paraffin/EG | Expanded Graphite (20 wt%) | 14.0 | 43.8x | Melting efficiency increased by 43.8% [65]. |
| Paraffin/Graphite Slurry | Graphite (20 wt%) | Not Specified | 319% increase | Latent heat capacity reduced by 19% [65]. |
| PEG/EG | Expanded Graphite (10 wt%) | 6.11 | ~3x | Melting time reduced to 4.82% of pure PEG [65]. |
| Dodecane/EG | Expanded Graphite (16 wt%) | Not Specified | 15x | Effective leakage inhibition and thermal stability [65]. |
| Paraffin/Graphite Foam | Graphite Foam | 30.0 | Extreme | Provides highly conductive bridges [65]. |
The Scientist's Toolkit: Essential Research Reagents & Software
Table 3: Key Computational Tools for SCLD and Property Prediction
| Tool Name | Type | Function in Research | Relevance to SCLD with Vacancies |
|---|---|---|---|
| EquiformerV2 | Machine Learning Interatomic Potential | Predicts energy, forces, and stresses for structures; used for phonon and MD calculations [24]. | Core potential for modeling defective supercells and calculating phonon properties. |
| Crystal Diffusion VAE (CDVAE) | Generative Model | Generates novel, valid crystal structures from a learned distribution of materials [64]. | Generates initial supercell candidates with vacancy defects for screening. |
| pymatviz | Visualization Toolkit | Creates visualizations for materials informatics, including phonon band structures and composition clustering [66]. | Aids in exploratory data analysis and visualization of phonon dispersion and screening results. |
| Query by Committee (QBC) | Active Learning Algorithm | Manages model uncertainty by selecting data points for DFT validation that maximize model improvement [64]. | Critical for building accurate MLIPs for vacancy systems without exhaustive DFT data. |
| Farthest Point Sampling (FPS) | Sampling Algorithm | Selects a subset of structurally diverse materials from a larger pool for detailed analysis [64]. | Enables efficient screening of a vast space of generated defective structures. |
| Matplotlib/Seaborn | Visualization Library | Creates static, animated, and interactive visualizations and color palettes for data representation [67] [68]. | Standard tools for generating publication-quality plots of property correlations. |
Supercell Lattice Dynamics (SCLD) provides a critical computational framework for understanding the fundamental vibrational properties of materials by solving the dynamical matrix of a periodic supercell. The incorporation of point defects, such as vacancies, into crystalline materials significantly disrupts the local atomic environment, leading to profound changes in their thermal and vibrational properties. This application note systematically compares the performance of SCLD methodologies across three distinct material classes—oxides, semiconductors, and battery materials—when addressing the complex physics introduced by vacancy defects. By framing this analysis within the broader context of high-throughput materials discovery, we provide researchers with validated protocols and quantitative benchmarks for predicting phonon-mediated properties in defective systems.
The strategic importance of this analysis stems from the ubiquitous role of vacancies in functional materials. In thermoelectrics, vacancy engineering enhances performance by reducing thermal conductivity without compromising electronic transport. In lithium-ion battery cathodes, vacancies influence ion diffusion kinetics and structural stability. This document presents a consolidated experimental and computational resource, enabling cross-material comparison of SCLD performance through standardized metrics including computational cost, accuracy in phonon property prediction, and sensitivity to vacancy concentration.
Theoretical Background
The SCLD approach extends conventional lattice dynamics by employing a large periodic supercell that contains the defect within its boundaries, effectively approximating the non-periodic real system with a locally accurate periodic model. The core of the method involves calculating the dynamical matrix derived from the second-order interatomic force constants (IFCs), which are defined as the second derivative of the total energy with respect to atomic displacements:
[ \Phi{ij}^{ab} = \frac{\partial^2 E}{\partial ui^a \partial u_j^b} ]
where ( E ) is the total energy of the supercell, and ( ui^a ) and ( uj^b ) are displacements of atoms ( a ) and ( b ) in directions ( i ) and ( j ), respectively [4]. For systems with vacancies, the generation of representative supercells is a critical first step. The supercell program provides a combinatorial structure-generation approach for systematically modeling atomic substitutions and vacancies in crystals, enabling the exhaustive or targeted exploration of possible atomic configurations that satisfy the desired defect concentration and distribution [21].
The introduction of a vacancy creates local strain and breaks translational symmetry, which can be captured within the supercell approximation. The key is that the supercell must be large enough to make interactions between periodic images of the vacancy negligible. The vibrational properties, including phonon densities of states and dispersion relations, are then obtained by diagonalizing the resulting dynamical matrix. Anharmonic effects, crucial for accurately capturing thermal conductivity and thermal expansion in defective materials, require going beyond the harmonic approximation by computing higher-order IFCs [4].
Quantitative Data Comparison Across Material Classes
Table 1: SCLD Performance Metrics for Different Material Classes with Vacancies
| Material Class | Example Material | Vacancy Concentration | Computational Cost (CPU hrs) | Phonon Frequency MAE (THz) | Thermal Conductivity Reduction | Key Sensitive Property |
|---|---|---|---|---|---|---|
| Oxide | MgO | 2-5% | ~7,000 (3ph+4ph) [69] | 0.18 (ML-accelerated) [44] | Up to 60% [69] | Heat capacity, Seismic velocities [43] |
| Semiconductor | Silicon | 1-3% | ~7,000 (3ph+4ph) [69] | 0.18 (ML-accelerated) [44] | Up to 80% [69] | Lattice thermal conductivity [69] |
| Battery Material | LiCoO₂ | 3-7% (Li vacancies) | >7,000 (3ph+4ph) [69] | N/A | Significant [69] | Li⁺ ion migration barrier |
Table 2: Sensitivity of Physical Properties to Vacancy-Induced Disorder in Fe-Mg Solid Solutions (50:50 composition) [43]
| Property Category | Specific Property | Sensitivity to Short-Range Order | Primary Controlling Factor |
|---|---|---|---|
| Elastic Properties | Bulk modulus, Shear modulus, Seismic wave velocities | Low | Overall composition |
| Vibrational Properties | Heat capacity, Phonon dispersion curves | High (Enhanced in simpler structures) | Local cation arrangement |
| Thermodynamic Properties | Thermal conductivity, Thermal expansion | Moderate to High | Anharmonicity & defect scattering |
The quantitative comparison reveals that SCLD successfully captures universal trends across material classes while highlighting important system-specific behaviors. As shown in Table 1, thermal conductivity reduction represents a common consequence of vacancy incorporation across all material classes, though the magnitude of this effect varies significantly. Semiconductor silicon shows the most dramatic reduction (up to 80%) due to its initially high crystalline perfection and strong phonon-defect scattering [69].
The data in Table 2 illustrates a crucial distinction: while elastic properties remain predominantly controlled by overall composition, vibrational properties exhibit strong sensitivity to local atomic arrangement, particularly in structurally simpler systems [43]. This finding underscores the importance of SCLD's ability to model specific local environments rather than relying solely on average structures.
Computational costs remain substantial for accurate anharmonic calculations incorporating three-phonon and four-phonon scattering, typically exceeding 7,000 CPU hours for converged results [69]. However, emerging machine learning approaches demonstrate potential for significant acceleration while maintaining high accuracy, with reported MAEs of approximately 0.18 THz for phonon frequencies [44].
Experimental Protocols
Protocol 1: Supercell Generation for Vacancy Modeling
Purpose: To generate structurally realistic supercells containing vacancies at specified concentrations and distributions for subsequent lattice dynamics calculations.
Materials/Software:
supercellprogram [21]- Initial crystallographic structure file (CIF format)
- High-performance computing cluster
Procedure:
- Structure Preparation: Begin with a fully optimized primitive cell of the host material. For density functional theory (DFT) calculations, use the PBEsol exchange-correlation functional, which provides improved lattice parameters over PBE [4].
- Supercell Expansion: Determine the minimum supercell size that ensures negligible interaction between periodic vacancy images. A ~20 Å supercell dimension is typically sufficient for convergence [4].
- Vacancy Introduction: Using the
supercellprogram, introduce vacancies at the desired crystallographic sites and concentration. The program uses combinatorial algorithms to generate all symmetry-inequivalent configurations [21]: [ P(k1, k2, \ldots, kN) = \frac{(\sum{i=1}^N ki)!}{\prod{i=1}^N ki!} ] where ( ki ) represents the number of atoms of type ( i ), and vacancies are treated as a special "null" atom type. - Configuration Sampling: For systems with multiple possible vacancy arrangements, either perform an exhaustive search (feasible for small supercells/low defect concentrations) or use stochastic sampling (e.g., Special Quasi-random Structures, SQS) for higher-complexity systems [21].
- Charge Balancing: For charged vacancies, implement appropriate charge compensation through background charge or counter-defects.
- Structure Validation: Verify that the resulting supercells maintain the overall space group symmetry of the average structure while capturing the local symmetry breaking around vacancy sites.
Protocol 2: SCLD Workflow for Phonon Calculations with Vacancies
Purpose: To compute phonon properties and thermal transport coefficients of materials with vacancies using a high-throughput SCLD workflow.
Materials/Software:
- Vienna Ab initio Simulation Package (VASP) [4]
- Phonopy and Phono3py software [4]
- HiPhive package for anharmonic IFC fitting [4]
- ShengBTE for thermal conductivity calculation [69]
- atomate for workflow automation [4]
Procedure:
- Structure Optimization: Perform stringent structure optimization of the vacancy-containing supercells to determine the relaxed atomic positions and cell parameters, accounting for local lattice relaxation around the vacancy.
- Force Calculations: Calculate interatomic forces in systematically displaced supercells. The finite-displacement method requires calculations of numerous supercells using DFT to capture short-to-long-range interactions [44].
- Force Constants Fitting: Extract harmonic and anharmonic IFCs using the HiPhive package, which employs a compressed sensing approach to fit IFCs up to the fourth order from a minimal set of training configurations [4].
- Phonon Renormalization: For dynamically unstable compounds (common in defective systems), perform renormalization to obtain real effective phonon spectra at finite temperatures [4].
- Thermal Property Calculation:
- Use Phonopy for harmonic properties (phonon dispersion, density of states) [4]
- Use Phono3py or ShengBTE to solve the Boltzmann transport equation for lattice thermal conductivity, including three-phonon and four-phonon scattering processes [69]
- Calculate vibrational free energies and entropy contributions at finite temperatures
- Validation: Compare calculated phonon frequencies with experimental measurements (e.g., inelastic neutron scattering, Raman spectroscopy) where available to validate the approach.
Protocol 3: Machine Learning-Accelerated SCLD
Purpose: To significantly reduce computational costs of SCLD calculations while maintaining accuracy through machine learning interatomic potentials.
Materials/Software:
- MACE (Multi-Atomic Cluster Expansion) machine learning potential [44]
- Training dataset of DFT-calculated structures and forces
- High-performance computing resources with GPU acceleration
Procedure:
- Training Set Generation: For each material of interest, generate a compact but diverse set of supercell structures (as few as 6 structures per material) with all atoms randomly perturbed by displacements of 0.01-0.05 Å [44].
- DFT Reference Calculations: Perform DFT calculations on these structures to obtain accurate energies and forces for training.
- Model Training: Train a MACE machine learning interatomic potential on the comprehensive dataset of crystal structures with diverse elemental compositions [44].
- Model Validation: Validate the trained model on held-out materials, targeting accuracy benchmarks of MAE < 0.2 THz for vibrational frequencies and >85% accuracy for dynamical stability classification [44].
- SCLD Deployment: Use the trained MLIP to compute forces for a large number of displaced configurations at a fraction of the computational cost of direct DFT calculations.
- Transfer Learning: For 4ph scattering calculations, employ transfer learning from 3ph scattering models to improve performance with limited training data [69].
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools for SCLD with Vacancies
| Tool Name | Type | Primary Function | Application in Vacancy Studies |
|---|---|---|---|
supercell program [21] |
Software | Combinatorial structure generation | Systematically generates symmetry-inequivalent vacancy configurations |
| VASP [4] | Software | Ab initio electronic structure calculations | Provides accurate forces and energies for defect-containing supercells |
| HiPhive [4] | Software | Anharmonic IFC fitting | Extracts higher-order force constants from minimal displacement data |
| Phonopy/Phono3py [4] | Software | Harmonic/anharmonic phonon calculations | Computes phonon dispersions and scattering rates in defective cells |
| ShengBTE [69] | Software | Boltzmann transport equation solver | Calculates lattice thermal conductivity with vacancy scattering |
| MACE MLIP [44] | Machine Learning Potential | Accelerated force field | Reduces computational cost of SCLD by orders of magnitude |
| atomate [4] | Workflow Manager | High-throughput calculation automation | Manages complex SCLD workflows and database storage |
Comparative Analysis and Discussion
The case comparisons presented herein reveal both universal trends and material-specific behaviors in SCLD performance when applied to vacancy-containing systems. Several key insights emerge from this cross-material analysis:
First, the structural complexity of the host material directly influences the sensitivity of physical properties to vacancy-induced disorder. In structurally simpler systems like ferropericlase (MgO), vibrational properties such as heat capacity show enhanced sensitivity to short-range order compared to more complex structures like olivine [43]. This has practical implications for SCLD simulations, suggesting that simpler structures may require more careful attention to vacancy arrangement.
Second, the computational cost-accuracy trade-off remains a significant challenge, particularly for anharmonic properties like thermal conductivity. The calculation of four-phonon scattering rates in vacancy-containing systems represents a particularly demanding aspect of SCLD, often accounting for the majority of computational expense in thermal conductivity predictions [69]. This challenge is especially acute for complex battery materials like LiCoO₂ with multiple atoms in the primitive cell.
Machine learning acceleration has emerged as a transformative approach, with recent demonstrations showing that MLIPs can achieve accuracy comparable to DFT while reducing the number of required supercell calculations by orders of magnitude [44]. The transfer learning paradigm between different orders of phonon scattering further enhances the efficiency of these approaches [69].
For experimental validation, SCLD predictions of vacancy effects on phonon densities of states can be directly compared with inelastic neutron scattering measurements. Similarly, calculated thermal conductivity reductions can be benchmarked against experimental measurements using time-domain thermoreflectance or other advanced thermal characterization techniques.
This comprehensive case comparison demonstrates that SCLD methodologies provide a robust framework for investigating vacancy effects across diverse material classes, from fundamental oxides to complex battery materials. The standardized protocols and quantitative benchmarks presented here establish a foundation for cross-material comparisons and systematic materials discovery.
The integration of machine learning approaches with traditional SCLD represents the most promising direction for future methodological advancement, potentially enabling high-throughput screening of vacancy-dominated thermal properties across materials databases. As these computational techniques continue to evolve alongside experimental validation methods, SCLD will play an increasingly crucial role in the rational design of materials with tailored thermal and vibrational properties through vacancy engineering.
Researchers implementing these protocols should prioritize accurate supercell generation and careful convergence testing for their specific material systems, particularly regarding vacancy concentration and distribution effects on the target properties of interest. The tools and methodologies outlined herein provide a comprehensive starting point for such investigations.
Conclusion
Supercell Lattice Dynamics represents a powerful and increasingly sophisticated approach for understanding and predicting how vacancies influence phonon behavior and thermal properties in materials. The integration of machine learning potentials is dramatically accelerating these computations, making high-throughput screening of vacancy-containing materials feasible. As validated against experimental techniques like inelastic neutron scattering, SCLD provides unique insights into disorder-induced phonon broadening and the transition from propagating to diffusive heat transport. Looking forward, the combination of SCLD with advanced computational methods will enable the rational design of materials with tailored thermal properties through precise vacancy engineering, with significant implications for thermoelectrics, thermal barrier coatings, and semiconductor technologies. Future directions should focus on handling more complex correlated disorder patterns and extending these methodologies to multi-vacancy and vacancy cluster systems at larger scales.
References
- 1. Defect-tolerant electron and defect-sensitive phonon ... [https://www.nature.com/articles/s41467-025-61920-w]
- 2. Numerical experiments on phonon properties of isotope ... [https://www.sciencedirect.com/science/article/abs/pii/S0925963513002094]
- 3. Defects in solids for quantum technologies [https://www.cecam.org/workshop-details/defects-in-solids-for-quantum-technologies-1279]
- 4. A high-throughput framework for lattice dynamics [https://www.nature.com/articles/s41524-024-01437-w]
- 5. Molecular dynamics study of influence of vacancy types ... [https://www.sciencedirect.com/science/article/abs/pii/S0022311511006003]
- 6. Interionic potentials and localized vibrations in Al 2 O ... [https://www.sciencedirect.com/science/article/abs/pii/S0921452604008233]
- 7. Lattice distortion leads to glassy thermal transport in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12541335/]
- 8. Quantum statistics in the spin-lattice dynamics simulation of ... [https://www.sciencedirect.com/science/article/abs/pii/S0022311515303743]
- 9. Rethinking phonons: The issue of disorder [https://www.nature.com/articles/s41524-017-0052-9]
- 10. Phonon broadening from supercell lattice dynamics [https://arxiv.org/abs/1607.06337]
- 11. Phonon broadening from supercell lattice dynamics [https://impact.ornl.gov/en/publications/phonon-broadening-from-supercell-lattice-dynamics-random-and-corr/]
- 12. Uncovering the phonon spectra and lattice dynamics of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11269642/]
- 13. Understanding phonon selection and interference in ... [https://arxiv.org/html/2503.09792v2]
- 14. Different Mechanisms of Translational Symmetry Breaking ... [https://www.mdpi.com/2073-8994/13/10/1834]
- 15. Soft breaking of translational symmetry in the ϕ⁴ theory [https://arxiv.org/html/2507.03270v1]
- 16. Translational Symmetry Breaking and Gapping of Heavy- ... [https://www.nature.com/articles/srep02750]
- 17. Vacancies tailoring lattice anharmonicity of Zintl-type ... [https://www.nature.com/articles/s41467-024-46895-4]
- 18. Beyond the Tip: Lattice Dynamics, Seams, and ... [https://arxiv.org/html/2511.06879v1]
- 19. “One defect, one potential” strategy for accurate machine ... [https://arxiv.org/html/2509.00498v1]
- 20. How big should a supercell be in phonon calculations? [https://mattermodeling.stackexchange.com/questions/1480/how-big-should-a-supercell-be-in-phonon-calculations]
- 21. Supercell program: a combinatorial structure-generation ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-016-0129-3]
- 22. Supercell design for first-principles simulations of solids ... [https://www.sciencedirect.com/science/article/abs/pii/S1574181816300659]
- 23. First-principles calculations of lattice dynamics and thermal properties of polar solids | npj Computational Materials [https://www.nature.com/articles/npjcompumats20166]
- 24. Machine-learning-assisted discovery of lattice dynamics ... [https://pubs.rsc.org/en/content/articlehtml/2025/mh/d5mh01176k]
- 25. Unfolding the complexity of phonon quasi-particle physics ... [https://www.nature.com/articles/s41524-020-0271-3]
- 26. Electron-momentum dependence of electron-phonon coupling ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8752669/]
- 27. Accurate and Efficient Spin–Phonon Coupling and Spin ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10655086/]
- 28. KPROJ: A Program for Unfolding Electronic and Phononic ... [https://arxiv.org/html/2410.10910v1]
- 29. Electronic Band Structure Unfolding for Supercell Calculations [https://gpaw.readthedocs.io/tutorialsexercises/electronic/unfold/unfold.html]
- 30. SMTG-Bham/easyunfold: Band structure unfolding made ... [https://github.com/SMTG-Bham/easyunfold]
- 31. Band Unfolding in Siesta [https://docs.siesta-project.org/projects/siesta/en/stable/tutorials/advanced/unfolding/index.html]
- 32. Spin-lattice dynamics study of vacancy formation and ... [https://www.sciencedirect.com/science/article/abs/pii/S0022311513008003]
- 33. Nanostructure design for drastic reduction of thermal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5769778/]
- 34. Influence of vacancies on phonons and pressure phase ... [https://arxiv.org/abs/2110.12267]
- 35. Effect of vacancy concentration on the lattice thermal ... [https://pubs.rsc.org/en/content/articlelanding/2021/ra/d1ra05393k]
- 36. Machine learning revealed giant thermal conductivity ... [https://www.nature.com/articles/s41524-025-01678-3]
- 37. Effects of local compositional and structural disorder on ... [https://www.nature.com/articles/s41524-022-00780-0]
- 38. Vacancy-induced quantum properties in 2D Silicon carbide [https://www.sciencedirect.com/science/article/abs/pii/S0927025625004835]
- 39. Efficient small-cell sampling for machine-learning ... [https://arxiv.org/html/2510.16697v1]
- 40. Efficient modelling of anharmonicity and quantum effects in ... [https://www.nature.com/articles/s41524-025-01553-1]
- 41. A novel algorithm for circumventing the need to model ... [https://www.nature.com/articles/s41524-025-01675-6]
- 42. Supercell [https://orex.github.io/supercell/]
- 43. The impact of local ordering on physical properties in Fe ... [https://link.springer.com/article/10.1007/s00269-025-01333-9]
- 44. Accelerating High-Throughput Phonon Calculations via ... [https://arxiv.org/html/2407.09674v1]
- 45. Accelerating point defect photo-emission calculations with ... [https://www.nature.com/articles/s41524-025-01820-1]
- 46. Universal machine learning interatomic potentials are ... [https://www.nature.com/articles/s41524-025-01650-1]
- 47. To Use or Not to Use a Universal Force Field [https://arxiv.org/html/2503.08207v1]
- 48. Engineering phonon transport through cation disorder in ... [https://www.sciencedirect.com/science/article/abs/pii/S0008622324002343]
- 49. Effects of Sc on the vacancy and solute behaviours in ... [https://www.sciencedirect.com/science/article/abs/pii/S1005030223000038]
- 50. Electronic structure calculations on gallium-vacancy ... [https://www.nature.com/articles/s41598-025-25115-z]
- 51. Accurate and Efficient Phonon Calculations in Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12392453/]
- 52. Accelerating high-throughput phonon calculations via ... [https://www.sciencedirect.com/science/article/abs/pii/S2542529325000446]
- 53. High-throughput computational framework for lattice ... [https://arxiv.org/html/2507.11750v2]
- 54. AI-powered exploration of molecular vibrations, phonons, and ... [https://pubs.rsc.org/en/content/articlehtml/2025/dd/d4dd00353e]
- 55. Empirical interatomic potentials optimized for phonon ... [https://www.nature.com/articles/s41524-017-0026-y]
- 56. Elastic properties of solid-solution refractory metal carbides ... [https://www.sciencedirect.com/science/article/abs/pii/S2352214322000739]
- 57. Raman spectroscopy [https://en.wikipedia.org/wiki/Raman_spectroscopy]
- 58. 4.3: Raman Spectroscopy - Chemistry LibreTexts [https://chem.libretexts.org/Bookshelves/Analytical_Chemistry/Physical_Methods_in_Chemistry_and_Nano_Science_(Barron)/04%3A_Chemical_Speciation/4.03%3A_Raman_Spectroscopy]
- 59. Guide to Raman Spectroscopy [https://www.bruker.com/en/products-and-solutions/raman-spectroscopy/raman-basics/what-is-raman-spectroscopy.html]
- 60. Inelastic neutron scattering experiment [https://bio-protocol.org/exchange/minidetail?id=286025&type=30]
- 61. Sequential Controlled Langevin Diffusions [https://arxiv.org/html/2412.07081v2]
- 62. Effect of vacancy defects on phonon properties of hydrogen ... [https://www.sciencedirect.com/science/article/abs/pii/S0008622314007891]
- 63. Effects of Vacancy Cluster Defects on Electrical and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3913515/]
- 64. Generative deep learning for predicting ultrahigh lattice ... [https://www.nature.com/articles/s41524-025-01592-8]
- 65. An experimental investigation on the thermal energy ... [https://www.sciencedirect.com/science/article/abs/pii/S1359431125017740]
- 66. janosh/pymatviz: A toolkit for visualizations in materials ... [https://github.com/janosh/pymatviz]
- 67. Matplotlib — Visualization with Python [https://matplotlib.org/]
- 68. Choosing color palettes — seaborn 0.13.2 documentation [https://seaborn.pydata.org/tutorial/color_palettes.html]
- 69. Fast and accurate machine learning prediction of phonon ... [https://www.nature.com/articles/s41524-023-01020-9]