Green-Kubo Modal Analysis (GKMA): A Unified Framework for Thermal Conductivity in Disordered Solids
This article provides a comprehensive exploration of Green-Kubo Modal Analysis (GKMA), a powerful computational formalism for calculating modal contributions to thermal conductivity in complex materials.
Green-Kubo Modal Analysis (GKMA): A Unified Framework for Thermal Conductivity in Disordered Solids
Abstract
This article provides a comprehensive exploration of Green-Kubo Modal Analysis (GKMA), a powerful computational formalism for calculating modal contributions to thermal conductivity in complex materials. Tailored for researchers and scientists, we cover the foundational theory behind GKMA and its key advantage: providing a unified framework applicable to amorphous solids, crystalline alloys, and polymers. The piece delves into methodological implementation, practical troubleshooting for molecular dynamics simulations, and rigorous validation against established methods. By synthesizing these aspects, this resource aims to equip professionals with the knowledge to apply GKMA for accurate thermal analysis in disordered materials, offering profound insights that challenge and expand the conventional phonon gas model.
Beyond the Phonon Gas Model: GKMA Foundations for Disordered Materials
Theoretical Foundation of the Green-Kubo Formalism
The Green–Kubo relations provide the exact mathematical framework for calculating transport coefficients from the microscopic dynamics of a system at equilibrium [1]. These relations connect a macroscopic transport coefficient to the time-integral of the autocorrelation function of a corresponding microscopic flux [2] [1].
For thermal conductivity, the Green-Kubo relation expresses the thermal transport coefficient tensor components ( \gamma_{\alpha\beta} ) as:
[ \gamma{\alpha\beta} = \int{0}^{\infty} \langle J{\alpha}(t) J{\beta}(0) \rangle dt = \int{0}^{\infty} C{\alpha\beta}(t) dt ]
where ( J{\alpha} ) and ( J{\beta} ) are components of the microscopic heat current vector, and ( C_{\alpha\beta}(t) ) is the current autocorrelation function (CAF) [2]. The angle brackets ( \langle \cdots \rangle ) denote the ensemble average in the equilibrium system.
This formalism is derived from the fluctuation-dissipation theorem, which fundamentally links the relaxation of perturbations to spontaneous fluctuations in equilibrium [1]. In practice, during molecular dynamics (MD) simulations, the continuous current becomes a discrete series, and the correlation function at time ( k\Delta t ) (where ( \Delta t ) is the MD time step) is calculated as:
[ C{k}^{\alpha\beta} = \frac{1}{N-k} \sum{i=0}^{N-k-1} J{i+k}^{\alpha} \cdot J{i}^{\beta} ]
where ( N ) represents the total number of steps in the simulation [2].
Table 1: Key Mathematical Quantities in Green-Kubo Formalism
| Symbol | Term | Physical Meaning |
|---|---|---|
| ( \gamma_{\alpha\beta} ) | Transport coefficient tensor | Macroscopic thermal conductivity |
| ( J_{\alpha} ) | Microscopic heat current | Atomic-level heat flow |
| ( C_{\alpha\beta}(t) ) | Current autocorrelation function (CAF) | Temporal correlation of heat currents |
| ( \langle J{\alpha}(t)J{\beta}(0) \rangle ) | Ensemble average | Equilibrium fluctuation average |
Practical Implementation and Protocols
Current Autocorrelation Function Calculation
In molecular dynamics simulations, the trajectory is typically divided into ( M ) intervals of equal length to improve statistics. The average CAF is then computed as:
[ C{k}^{\alpha\beta} = \frac{1}{M} \sum{A=1}^{M} \langle J^{\alpha} \cdot J^{\beta} \rangle_{k}^{(A)} ]
where the index ( A ) runs over the different intervals [2]. The statistical uncertainty of the CAF represents a critical consideration:
[ u(C{k}^{\alpha\beta}) = \frac{\sigma{k}^{\alpha\beta}}{\sqrt{M(N-k)}} = \frac{1}{\sqrt{M(N-k)-1}} \left[ \frac{1}{M} \sum{A=1}^{M} \langle (J^{\alpha})^{2} \cdot (J^{\beta})^{2} \rangle{k}^{(A)} - (C_{k}^{\alpha\beta})^{2} \right]^{1/2} ]
where ( \sigma_{k} ) is the standard deviation of the ( k )-th value of the CAF [2]. This uncertainty quantification enables researchers to assess the reliability of their calculations.
Running Integral and Transport Coefficient Estimation
The running integral of the CAF provides the cumulative estimate of the transport coefficient up to time ( k\Delta t ). Using the trapezoidal integration scheme:
[ I{k}^{\alpha\beta} = \frac{\Delta t}{2} \sum{i=0}^{k} (C{i}^{\alpha\beta} + C{i+1}^{\alpha\beta}) ]
The uncertainty of this running integral propagates from the CAF uncertainties:
[ u(I{k}^{\alpha\beta}) = \frac{\Delta t}{2} \sqrt{ \sum{i=0}^{k} \left[ u^{2}(C{i}^{\alpha\beta}) + u^{2}(C{i+1}^{\alpha\beta}) \right] } ]
This uncertainty grows approximately as ( \sqrt{k} ) with increasing time, creating the central challenge in Green-Kubo analysis: identifying the plateau region before noise dominates [2].
Graph 1: Green-Kubo Thermal Conductivity Calculation Workflow. The process begins with equilibrium molecular dynamics, proceeds through correlation analysis, and culminates in plateau identification for the final thermal conductivity value.
Advanced Analysis with KUTE Algorithm
The KUTE (green-Kubo Uncertainty-based Transport properties Estimator) algorithm addresses the challenge of arbitrary integration cutoffs by implementing a weighted averaging approach [2]. This method defines a running transport coefficient:
[ \gamma{i}^{\alpha\beta} = \frac{ \sum{k=i}^{N} I{k}^{\alpha\beta} / u^{2}(I{k}^{\alpha\beta}) }{ \sum{k=i}^{N} u^{-2}(I{k}^{\alpha\beta}) } ]
with statistical uncertainty:
[ u(\gamma{i}^{\alpha\beta}) = \frac{1}{N-i} \sqrt{ \frac{ \sum{k=i}^{N} (\gamma{i}^{\alpha\beta} - I{k}^{\alpha\beta})^{2} / u^{2}(I{k}^{\alpha\beta}) }{ \sum{k=i}^{N} u^{-2}(I_{k}^{\alpha\beta}) } } ]
When this sequence exhibits a plateau across a range of averaging origins, the plateau value is identified as the transport coefficient ( \gamma^{\alpha\beta} ) [2]. This approach systematically eliminates the need for arbitrary cutoffs that could alter results.
For the average isotropic thermal conductivity, the running average is computed as:
[ \gamma{i} = \frac{ \sum{\alpha} \gamma{i}^{\alpha\alpha} / u^{2}(\gamma{i}^{\alpha\alpha}) }{ \sum{\alpha} u^{-2}(\gamma{i}^{\alpha\alpha}) } ]
with corresponding uncertainty [2].
Application to Disordered Solids and GKMA Context
The Green-Kubo Modal Analysis (GKMA) method extends the standard Green-Kubo formalism to enable direct calculation of individual phonon mode contributions to thermal conductivity [3]. This approach provides a unified framework applicable to diverse material systems, including amorphous materials, crystalline solids, crystalline alloys, and polymers.
Within the context of disordered solids research, GKMA has revealed limitations of the conventional phonon gas model, demonstrating that certain vibrational modes exhibit behaviors not captured by traditional models [3]. This insight is particularly valuable for understanding the reduced thermal conductivity observed in disordered materials.
Table 2: Current Green-Kubo Methodologies for Thermal Transport
| Method | Key Principle | Advantages | Limitations |
|---|---|---|---|
| Standard Green-Kubo | Direct integration of heat current autocorrelation | No external perturbation needed; well-established | Statistical noise; arbitrary cutoff selection |
| KUTE Algorithm | Uncertainty-weighted averaging of running integral | Eliminates arbitrary cutoffs; robust error estimation | Python implementation required [2] |
| Cepstral Analysis | Denoising of heat flux power spectrum | Objective criteria for analysis; handles noise | Underestimates high thermal conductivity [4] |
| GKMA | Modal decomposition of thermal transport | Reveals individual mode contributions; universal formalism | Increased computational complexity [3] |
Research Reagent Solutions and Computational Tools
Table 3: Essential Computational Tools for Green-Kubo Simulations
| Tool/Software | Function | Application Context |
|---|---|---|
| KUTE Python Package | Transport properties estimation from MD | Implements uncertainty-based analysis [2] |
| OpenMM | Molecular dynamics engine | Polarizable MD simulations [2] |
| MACE MLIP | Machine-learning interatomic potential | Accelerated first-principles MD [4] |
| PACKMOL | Initial structure generation | Simulation box preparation [2] |
| CL&Pol Force Field | Polarizable force field | Ionic liquid simulations [2] |
Data Analysis and Uncertainty Quantification
The statistical uncertainty in Green-Kubo analysis grows with correlation time, making uncertainty quantification essential for reliable results. The KUTE algorithm implements a sophisticated approach where the plateau identification becomes objective through statistical weighting [2].
For complex systems like disordered solids, the convergence of Green-Kubo integrals may require extended simulation times. Recent approaches combine multiple strategies, including cepstral analysis and machine-learning potentials, to accelerate calculations while maintaining accuracy [4].
Graph 2: KUTE Algorithm Uncertainty-Based Analysis Protocol. This flowchart illustrates the iterative process of plateau identification in the KUTE algorithm, where statistical weighting eliminates subjective cutoff selection.
Experimental Protocol for Disordered Solids
System Preparation and Equilibration
Initial Structure Generation: Use PACKMOL or similar tools to create simulation cells of disordered solids with appropriate composition and density [2].
Energy Minimization: Perform energy minimization with a tolerance of 10 kJ/mol to remove high-energy configurations [2].
Stabilization Protocol:
- Run NpT ensemble simulations for 10+ ns to equilibrate density
- Follow with NVT ensemble simulations for 5+ ns at target temperature
- Use Langevin thermostat or Nosé-Hoover chains for temperature control
Production Simulation: Conduct extended NVT ensemble simulations (50+ ns) for correlation function calculation [2].
Heat Current Calculation and Analysis
Current Sampling: Calculate microscopic heat current at regular intervals (1-10 fs) during production simulation.
Correlation Function Computation: Divide trajectory into multiple intervals for ensemble averaging of current autocorrelation functions [2].
Running Integral Calculation: Apply numerical integration with uncertainty propagation at each time point.
Plateau Identification: Use KUTE algorithm or similar statistical methods to identify the converged thermal conductivity value from the plateau region of the running integral [2].
This protocol provides a robust framework for applying Green-Kubo formalism to disordered solids, particularly within the research context of GKMA, enabling insights into the fundamental thermal transport mechanisms in these complex materials.
The Limitation of Conventional Methods for Disordered Systems
Predicting the thermal conductivity of disordered solids, such as amorphous silicon dioxide (a-SiO₂), presents a significant challenge in materials science. While the phonon gas model (PGM) and related methods have been highly successful for crystalline materials, their application to amorphous systems is fundamentally limited [5]. This document outlines these limitations and introduces the Green-Kubo Modal Analysis (GKMA) method as a unified formalism that overcomes these challenges, providing detailed protocols for its application in thermal conductivity research.
Limitations of Conventional Methods
The primary conventional methods for assessing thermal conductivity face specific theoretical and practical hurdles when applied to disordered systems.
2.1 The Phonon Gas Model (PGM) and Its Shortcomings The PGM describes thermal conductivity (κ) as a function of phonon contributions to specific heat (Cν), phonon group velocities (v), and phonon mean free paths (MFPs) [5]. For crystalline compounds, periodicity allows for a straightforward definition of phonon dispersion and group velocity. However, in amorphous materials, the lack of periodicity means one cannot rigorously define the group velocity (dω/dk), making the application of the PGM highly questionable [5]. Consequently, MFP-based explanations of phonon transport become difficult to rationalize, as the majority of vibrational modes in amorphous materials are non-propagating (e.g., diffusons and localized modes) [5].
2.2 The Allen-Feldman (A-F) Method and Its Limitations The seminal A-F method addressed the PGM's velocity problem by expressing the thermal conductivity of amorphous materials based on a mode diffusivity instead of MFP [5]. However, a key limitation is that it does not fully include the effects of anharmonicity in atomic interactions. The only temperature dependence in the A-F model comes from the temperature-dependent specific heat, leading to predictions of essentially constant thermal conductivity above 200 K for a-SiO₂, which fails to capture the experimentally observed increase in thermal conductivity beyond room temperature [5].
2.3 Quantitative Data: Thermal Conductivity of Common Materials The table below illustrates the range of thermal conductivities for various materials, highlighting the low conductivity typical of disordered and amorphous systems compared to ordered crystals.
Table 1: Thermal Conductivity of Selected Materials at 25°C [6] [7]
| Material | State / Type | Thermal Conductivity (W/m·K) |
|---|---|---|
| Diamond | Crystalline Solid | 1000 |
| Copper | Metallic Crystal | 200 |
| Silicon | Crystalline Solid | 149 |
| Aluminum | Metallic Crystal | 226 |
| Alumina (Al₂O₃) | Crystalline Ceramic | 30-36 |
| Glass | Amorphous Solid | ~1.05 |
| Concrete | Disordered Composite | 0.13 - 1.80 |
| Polyurethane Foam | Disordered Polymer | 0.03 - 0.06 |
| Air | Gas | 0.026 |
| Amorphous Silicon Dioxide (a-SiO₂) | Amorphous Solid | ~1.4 (at 400K) |
Green-Kubo Modal Analysis (GKMA): A Unified Formalism
The GKMA method is a formalism that enables the direct calculation of individual phonon or normal mode contributions to thermal conductivity, applicable to crystalline solids, amorphous materials, polymers, and molecules within a single unified framework [3].
3.1 Theoretical Foundation The GKMA method combines the Green-Kubo (GK) formula with modal information from lattice dynamics [5]. The thermal conductivity is given by the integral of the heat current autocorrelation function: [ \kappa = \frac{1}{kB T^2 V} \int0^{\infty} \langle J(t) \cdot J(0) \rangle dt ] where ( kB ) is Boltzmann's constant, ( T ) is temperature, ( V ) is volume, and ( J ) is the heat current vector. The innovation of GKMA is its ability to decompose the total heat current ( J ) into contributions from individual normal modes ( Jn ), allowing the thermal conductivity to be expressed as a sum of modal contributions ( \kappa^{(n)} ) and mode-mode cross-correlations [5].
3.2 Key Insights from GKMA on Disordered Systems Applying GKMA to a-SiO₂ has yielded critical insights that challenge conventional understanding. Previously, localized modes (locons) were thought to have a negligible contribution to thermal conductivity due to their highly localized nature. However, GKMA results indicate that in a-SiO₂, locons contribute more than 10% to the total thermal conductivity between 400 K and 800 K and are largely responsible for the increase in thermal conductivity of a-SiO₂ above room temperature [5]. This effect, which cannot be explained by the PGM or the A-F method, offers a new perspective on phonon transport in glasses.
Experimental Protocols: GKMA Workflow and Methodology
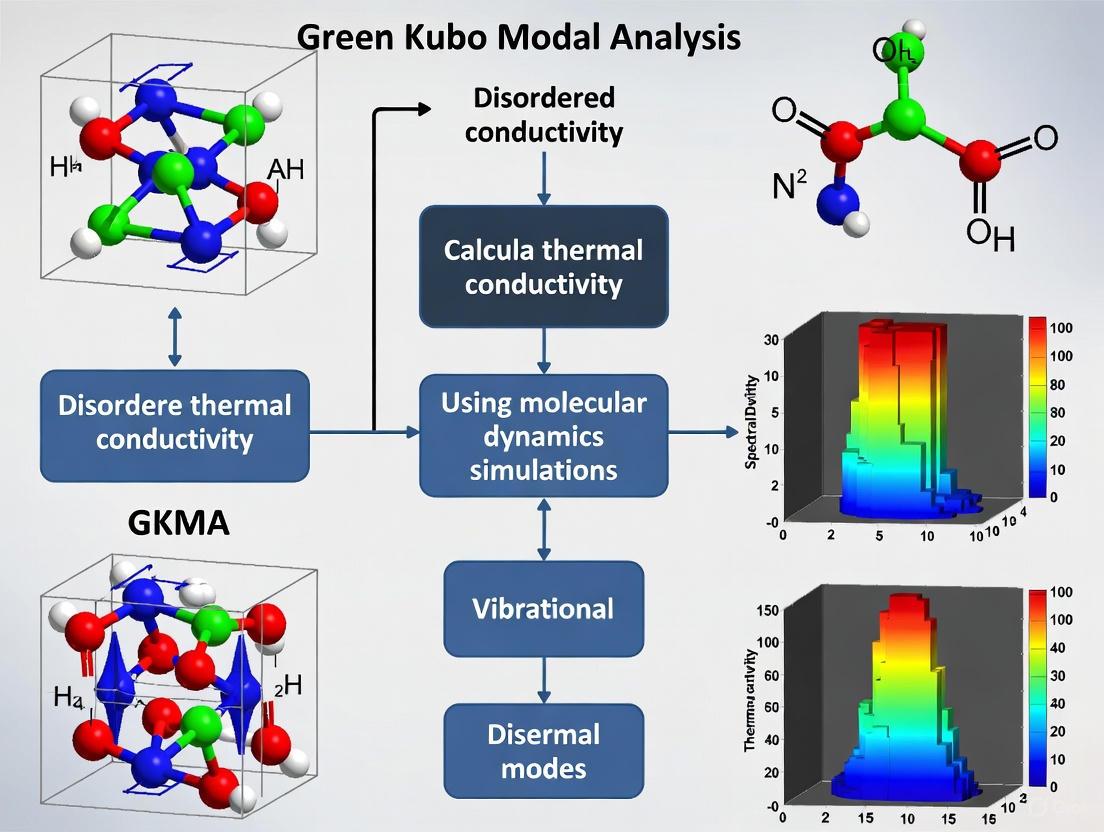
Protocol 1: GKMA Calculation Workflow for Amorphous Solids
This protocol details the step-by-step procedure for calculating the modal thermal conductivity of a material like a-SiO₂ using the GKMA method [5].
- System Preparation: Generate an atomic supercell of the amorphous material using ab initio or classical force field methods. Ensure the structure is relaxed to a stable equilibrium.
- Lattice Dynamics Calculation: Perform a lattice dynamics calculation on the supercell at the gamma point to compute the normal mode eigenvectors and frequencies.
- Equilibrium Molecular Dynamics (MD): Run a classical equilibrium MD simulation (NVE or NVT ensemble) of the same supercell for a sufficient duration to ensure proper sampling.
- Modal Projection: Project the atomic velocities from the MD trajectory onto the normal mode eigenvectors to obtain the time history of each normal mode's amplitude.
- Modal Heat Flux Calculation: Substitute the modal components of the atomic velocities into the heat flux operator to obtain each mode's instantaneous contribution to the heat flux.
- Green-Kubo Integration: For each mode, calculate its contribution to the thermal conductivity by integrating the autocorrelation of its heat flux contribution (including cross-correlations with other modes) using the Green-Kubo formula.
- Quantum Correction: Apply a quantum specific heat correction to the MD-derived modal contributions to account for proper quantum occupations at different temperatures. The corrected conductivity for a mode is: [ \kappa^{(n)}{corrected}(T) = fQ(\omegan, T) \times f\kappa^{(n)}(T) ] where ( f_Q ) is the ratio of quantum to classical specific heat [5].
The following diagram illustrates the computational workflow.
Protocol 2: Handling Statistical Uncertainty in Green-Kubo Calculations
A significant challenge in using GK relations is the statistical uncertainty in the integral of the current autocorrelation function (CAF). The kute (Green-Kubo Uncertainty-based Transport properties Estimator) Python package provides a robust methodology to address this [2].
- Current Autocorrelation Function (CAF) Calculation: Compute the CAF, ( C_{αβ}(t) ), from the microscopic current ( J ) obtained from an equilibrium MD simulation. For better statistics, the trajectory can be divided into multiple blocks.
- Uncertainty Quantification: Calculate the statistical uncertainty ( u(C_{αβ}) ) for each point of the CAF using the standard deviation across blocks.
- Running Integral & Uncertainty Propagation: Compute the running integral ( Ik ) of the CAF and propagate the uncertainty to obtain ( u(Ik) ), which grows with time.
- Weighted Averaging of the Plateau: Identify the plateau region of the running integral. Instead of a simple average, calculate a weighted average of the running integral values across the plateau, where the weights are the inverse squares of their uncertainties ( 1/u^2(I_k) ). This ensures data points with higher statistical significance contribute more to the final estimate of the transport coefficient [2].
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools and Methods for Thermal Transport Research
| Item/Reagent | Function/Description | Application Note |
|---|---|---|
| Molecular Dynamics (MD) Engine (e.g., LAMMPS, OpenMM) | Software to perform classical MD simulations, generating trajectories of atomic motions. | OpenMM was used for polarizable simulations of ionic liquids [2]. Essential for steps 3 and 4 in Protocol 1. |
| Green-Kubo Modal Analysis (GKMA) | A formalism to decompose thermal conductivity into individual mode contributions. | Provides unparalleled insight into the contributions of propagons, diffusons, and locons in disordered systems [5] [3]. |
| kute Python Package | An implementation for estimating transport coefficients from GK relations with integrated uncertainty quantification. | Eliminates the need for arbitrary cutoffs in the GK integral, providing more reliable and accurate results [2]. |
| Polarizable Force Fields (e.g., CL&Pol) | Advanced interatomic potentials that account for electronic polarization effects. | Crucial for accurate simulation of ionic systems like protic ionic liquids, leading to better agreement with experiment [2]. |
| Lattice Dynamics Code | Software to calculate vibrational normal modes and frequencies of an atomic structure. | Required for the initial modal analysis in the GKMA workflow (Step 2, Protocol 1) [5]. |
Visualization of Mode Contributions
The following diagram illustrates the distinct types of vibrational modes in amorphous materials and their contributions to heat transport as revealed by GKMA.
Core Principles of Green-Kubo Modal Analysis (GKMA)
Green-Kubo Modal Analysis (GKMA) is a formal methodology that enables the direct calculation of individual phonon or normal mode contributions to the thermal conductivity of materials. Its primary advancement is its generality; it can be applied to a wide range of materials, including crystalline solids, amorphous materials, random alloys, polymers, and even molecules, within a single, unified formalism [3] [8] [9]. This universality stands in contrast to the traditional Phonon Gas Model (PGM), which is best rationalized for crystalline materials and often fails to fully capture the thermal transport mechanisms in disordered systems [3] [10]. GKMA provides a framework for understanding thermal transport not through the lens of particle-like phonon scattering, but through the analysis of mode-mode correlations, offering new physical insights into the nature of heat conduction [8] [9].
Theoretical Foundations
The GKMA method synergistically combines the formalism of lattice dynamics with the Green-Kubo formula for thermal conductivity [8] [9]. The foundational equation for the thermal conductivity (( \kappa )) in the Green-Kubo approach is derived from equilibrium molecular dynamics (EMD) and is given by:
[ \kappa{xx} = \frac{1}{kB T^2 V} \int0^{\infty} \langle Jx(0) J_x(t) \rangle dt ]
Here, ( kB ) is Boltzmann’s constant, ( T ) is the absolute temperature, ( V ) is the system volume, ( Jx(t) ) is the heat flux vector in the x-direction at time ( t ), and the angle brackets represent the ensemble average of the heat flux autocorrelation function (HACF) [11].
The key innovation of GKMA is the decomposition of the total heat flux into contributions from individual vibrational modes. The atom velocities obtained from molecular dynamics simulations are projected onto the normal mode basis obtained from lattice dynamics [10]. For a system with ( N ) atoms, the velocity of atom ( i ) is:
[ \boldsymbol{v}i (t) = \frac{1}{\sqrt{mi}} \sum{n=1}^{3N} \boldsymbol{e}{i,n} \dot{X}_n(t) ]
where ( mi ) is the atomic mass, ( \boldsymbol{e}{i,n} ) is the eigenvector for atom ( i ) in mode ( n ), and ( \dot{X}n(t) ) is the time derivative of the normal mode coordinate for mode ( n ) [11]. The modal heat current ( \boldsymbol{J}n ) is then derived as [11]:
[ \boldsymbol{J}n = \left( \sum{i} \frac{1}{\sqrt{mi}} \mathbf{W}i \cdot \boldsymbol{e}{i,n} \right) \cdot \dot{X}n(t) ]
In this equation, ( \mathbf{W}_i ) is the per-atom virial tensor. Finally, the thermal conductivity contribution of a specific mode ( n ) is calculated using the GKMA formula [11] [10]:
[ \kappa{xx, n}(t) = \frac{1}{kB T^2 V} \int0^{t} \langle J{x, n}(t') J_{x}(0) \rangle dt' ]
This formulation allows the total thermal conductivity to be expressed as a direct summation of modal contributions (( \kappa = \sumn \kappan )), without the need to define a group velocity for each mode, making it particularly powerful for studying disordered materials where the concept of group velocity becomes ambiguous [8] [9].
Advantages of GKMA in Disordered Solids Research
The application of GKMA to disordered solids reveals physical insights that challenge or extend the conventional phonon gas model. The table below summarizes the key advantages of GKMA for this class of materials.
Table 1: Key Advantages of GKMA for Disordered Solids Research
| Feature | Implication for Disordered Solids Research |
|---|---|
| General Formalism | Provides a unified framework applicable to amorphous materials, random alloys, and crystals alike [3] [9]. |
| No Group Velocity Requirement | Bypasses the need to define a phonon velocity, which is problematic for non-propagating modes (diffusons, locons) in disordered systems [8] [9]. |
| Full Anharmonicity | Naturally includes anharmonic effects to full order through the use of molecular dynamics trajectories [9]. |
| Mode-Mode Correlations | Reveals the nature of phonon-phonon interactions via cross-correlations between different modes, moving beyond the scattering picture [8] [10]. |
| Quantification of "Optical-like" Mode Contributions | Enables direct calculation of the significant thermal conductivity contributions from negative phase quotient modes, which are often prominent in disordered solids [10]. |
A critical insight from GKMA studies is the important role played by vibrations with a negative Phase Quotient (PQ). The PQ measures the extent to which an atom and its nearest neighbors move in the same (positive PQ, acoustic-like) or opposing (negative PQ, optical-like) directions [10]. In crystalline materials, optical modes typically contribute minimally to thermal conductivity. However, GKMA has demonstrated that in disordered solids like amorphous SiO₂ (a-SiO₂) and amorphous carbon (a-C), a substantial portion of thermal conductivity can come from negative PQ, "optical-like" modes [10]. This highlights a fundamental difference in heat conduction mechanisms between ordered and disordered structures and underscores the value of GKMA in developing a more complete physical picture.
Experimental Protocols and Workflow
The following diagram illustrates the standard workflow for performing thermal conductivity analysis using the Green-Kubo Modal Analysis method.
Graph 1: Green-Kubo Modal Analysis Workflow. The process begins with harmonic lattice dynamics, proceeds through molecular dynamics simulation, and culminates in the calculation of mode-specific thermal conductivity.
Protocol Steps
Supercell Lattice Dynamics (SCLD)
- Purpose: To obtain the harmonic frequencies and eigenvectors (normal modes) of the atomic supercell.
- Procedure: Perform a dynamical matrix diagonalization on an equilibrated atomic structure. This step identifies all ( 3N ) vibrational modes of the system [10].
Molecular Dynamics (MD) Simulation
- Purpose: To generate a realistic, anharmonic trajectory of the atomic motions at the desired temperature.
- Procedure: Run an equilibrium MD simulation using an appropriate ensemble (e.g., NVT) and a reliable interatomic potential. The simulation should be sufficiently long to ensure proper sampling of the phase space and convergence of the heat flux autocorrelation [11] [10].
Velocity Projection
- Purpose: To decompose the atomic velocities from the MD trajectory into their modal contributions.
- Procedure: At multiple time steps throughout the MD trajectory, project the atomic velocities ( \boldsymbol{v}i(t) ) onto the normal mode basis ( \boldsymbol{e}{i,n} ) using the transformation ( \dot{X}n(t) = \sum{i} \sqrt{mi} \boldsymbol{e}{i,n} \cdot \boldsymbol{v}_i(t) ) [11] [10].
Modal Heat Current Calculation
- Purpose: To compute the heat flux carried by each individual mode.
- Procedure: Using the projected modal velocities ( \dot{X}n(t) ) and the per-atom virial ( \mathbf{W}i ), calculate the instantaneous modal heat current ( \boldsymbol{J}_n(t) ) for each mode [11].
Green-Kubo Integration
- Purpose: To calculate the contribution of each mode to the thermal conductivity.
- Procedure: For each mode ( n ), evaluate the cross-correlation between its heat current ( J{x, n}(t) ) and the total heat current ( J{x}(0) ) at time zero. Integrate this correlation function over time to obtain ( \kappa_{xx, n} ) [11] [10].
The Scientist's Toolkit
Successful application of GKMA relies on a combination of software tools, computational methods, and analytical concepts. The following table details these essential "research reagents."
Table 2: Essential Research Reagents and Tools for GKMA
| Tool Category | Specific Item/Concept | Function in GKMA Analysis |
|---|---|---|
| Software & Codes | LAMMPS | A widely used MD simulator; can be customized for heat flux breakdown and GKMA calculations [12]. |
| GPUMD | A molecular dynamics code with built-in functionalities for GKMA and other advanced heat transport methods [11]. | |
| Computational Methods | Supercell Lattice Dynamics (SCLD) | Calculates all vibrational modes (frequencies and eigenvectors) of an atomic supercell, providing the modal basis [10]. |
| Equilibrium MD (EMD) | Generates the atomic trajectory used to compute the anharmonic heat flux and its correlations [11]. | |
| Green-Kubo Formula | The foundational relation connecting the heat flux autocorrelation function to macroscopic thermal conductivity [12] [11]. | |
| Analytical Frameworks | Phonon Gas Model (PGM) | The conventional theory for crystalline materials; serves as a benchmark for contrasting GKMA findings [3] [10]. |
| Phase Quotient (PQ) | A metric to classify modes as acoustic-like (positive PQ) or optical-like (negative PQ), crucial for interpreting results in disordered solids [10]. | |
| PDL Classification | Categorizes modes as Propagons, Diffusons, or Locons; GKMA can quantify the thermal conductivity from each category [10]. |
Data Presentation and Analysis
Applying GKMA to different material classes reveals distinct patterns in how vibrational modes contribute to heat transport. The table below synthesizes key findings from GKMA studies, highlighting the contrast between ordered and disordered materials.
Table 3: Modal Contributions to Thermal Conductivity in Different Materials via GKMA
| Material System | Material Type | Key GKMA Findings on Modal Contributions |
|---|---|---|
| Crystalline Silicon | Crystalline | Optical phonon contributions are small (~5% at room temperature), consistent with the phonon gas model [10]. |
| Amorphous Silicon (a-Si) | Amorphous | GKMA predictions of temperature-dependent thermal conductivity show excellent agreement with experimental data [9]. |
| Amorphous SiO₂ (a-SiO₂) | Amorphous | Vibrations with negative Phase Quotient ("optical-like" modes) make a significant contribution to the overall thermal conductivity [10]. |
| Amorphous Carbon (a-C) | Amorphous | Similar to a-SiO₂, a substantial portion of heat is conducted by negative Phase Quotient modes [10]. |
| InₓGa₁₋ₓAs Alloy | Random Alloy | Modes with negative Phase Quotient contribute non-trivially to thermal conduction, deviating from pure crystal behavior [10]. |
The data in Table 3 underscores a central conclusion enabled by GKMA: the physical picture of thermal transport in disordered solids is fundamentally different from that in crystals. The significant role of "optical-like" modes (negative PQ) in disordered materials necessitates a move beyond the traditional phonon gas model. GKMA provides the unified formalism required to quantify these contributions and develop a more general understanding of heat conduction.
Thermal conductivity management is a cornerstone for advancing technology, from consumer electronics to sustainable energy solutions. For decades, the phonon gas model (PGM) has been the dominant theoretical framework for understanding heat conduction in solids [13]. This model treats phonons—quantized lattice vibrations—as a gas of particles, with thermal conductivity (κ) described by the familiar relationship κ = (1/3)ΣcᵥvᵥΛᵥ, which sums the contributions of individual phonon modes based on their specific heat (cᵥ), group velocity (vᵥ), and mean free path (Λᵥ) [14]. While this approach has proven remarkably successful for crystalline materials, its application to disordered solids like amorphous silicon dioxide (a-SiO₂) presents fundamental challenges [15] [13].
The core limitation of the PGM emerges in disordered systems where the lack of periodicity prevents rigorous definition of phonon velocities and wave vectors—properties inherent to the model's foundation [13]. This theoretical gap has created a critical need for a more comprehensive framework capable of describing thermal transport across all material classes. The Green-Kubo Modal Analysis (GKMA) method addresses this need by providing a unified formalism that enables direct calculation of individual phonon contributions to thermal conductivity for both ordered and disordered materials [3] [15]. By moving beyond the constraints of the PGM, GKMA offers researchers a powerful tool to explore thermal transport mechanisms in complex material systems that have previously resisted accurate modeling.
Theoretical Foundation of GKMA
The Green-Kubo Modal Analysis method represents a significant advancement in computational materials science by combining the rigorous statistical mechanics of the Green-Kubo formula with detailed modal information derived from lattice dynamics calculations [15]. The fundamental theoretical breakthrough of GKMA lies in its ability to decompose the total thermal conductivity into exact contributions from individual vibrational modes, expressed as:
κ = Σκ₍ₙ₎
where κ₍ₙ₎ represents the thermal conductivity contribution of the n-th normal mode [15]. This decomposition is achieved through a sophisticated mathematical framework that projects atomic velocities from molecular dynamics simulations onto normal mode eigenvectors, enabling the calculation of mode-specific heat currents [15].
Unlike the PGM, GKMA does not rely on ill-defined phonon velocities or mean free paths in disordered systems. Instead, it computes thermal transport properties directly from the anharmonic interactions between atoms, captured through the time correlation of heat fluxes [15] [16]. This approach naturally incorporates all orders of anharmonicity, providing a more complete physical picture of thermal transport, particularly in amorphous materials where multi-phonon scattering events dominate [15]. The method's theoretical robustness stems from its foundation in the Green-Kubo formula, which connects microscopic fluctuations to macroscopic transport coefficients through the fluctuation-dissipation theorem, while the modal analysis component preserves the critical connection to the vibrational characteristics of the material [16].
Table: Core Mathematical Expressions in the GKMA Formalism
| Mathematical Expression | Physical Significance | Advantage Over PGM |
|---|---|---|
| ( \kappa = \sumn \kappa{(n)} ) | Decomposes total thermal conductivity into individual mode contributions [15] | Enables precise identification of heat-carrying modes |
| ( \vec{J} = \sumn \vec{J}n ) | Partitions total heat flux into modal heat fluxes [15] | Accounts for collective atomic vibrations rather than independent phonons |
| ( \kappa{(n)} = \frac{1}{kB T^2 V} \int0^\infty \langle \vec{J}n(t) \cdot \vec{J}_n(0) \rangle dt ) | Green-Kubo relation applied to each mode's heat flux [15] | Directly computes κ from dynamics, avoiding assumptions about velocity and relaxation time |
Quantitative Applications and Material Comparisons
The power of the GKMA methodology is demonstrated through its successful application across diverse material systems, providing unprecedented insights into thermal transport phenomena. In a landmark study of amorphous silicon dioxide (a-SiO₂), GKMA calculations revealed exceptional agreement with experimental thermal conductivity data across a temperature range from 400 K to 800 K [15]. This study yielded the surprising discovery that localized modes (locons), previously considered negligible for heat conduction, contribute more than 10% to the total thermal conductivity in this temperature regime [15]. This finding directly challenges conventional wisdom and demonstrates GKMA's ability to uncover previously overlooked thermal transport mechanisms.
For crystalline materials, GKMA maintains strong predictive capability while providing deeper mechanistic insights. Applications to crystalline silicon (c-Si) have shown excellent agreement with both experimental data and established modal analysis methods [15]. The method's versatility extends beyond simple elemental and binary systems, successfully predicting thermal properties in complex structures including crystalline alloys and even individual polymer chains where traditional methods struggle [15]. This cross-material applicability highlights GKMA's unique strength as a unified framework that transcends traditional material classifications.
Table: GKMA Thermal Conductivity Predictions Across Material Classes
| Material System | Key GKMA Prediction | Temperature Range | Agreement with Experiment |
|---|---|---|---|
| Amorphous Silicon Dioxide (a-SiO₂) | Locons contribute >10% to total κ [15] | 400 K - 800 K | Excellent [15] |
| Crystalline Silicon (c-Si) | Accurate modal contributions [15] | Room Temperature | Excellent [15] |
| Single Polythiophene Chains | Possible divergent κ in certain chain lengths [16] | Simulation Conditions | Further validation needed |
| Amorphous Silicon (a-Si) | Identifies non-PGM transport mechanisms [13] | 30 K - 1200 K | Good with quantum correction [13] |
Experimental Protocols and Computational Methodologies
GKMA Implementation Workflow
Implementing GKMA requires a structured computational workflow that integrates lattice dynamics with molecular dynamics simulations. The following protocol outlines the key steps for applying GKMA to amorphous material systems:
System Preparation: Construct an atomic supercell of the amorphous material using experimental data or melt-quench procedures. For a-SiO₂, this typically involves generating a sufficiently large model (containing several hundred atoms) with density and bond-angle distributions matching experimental characterization data [15].
Lattice Dynamics Calculation: Perform a normal mode analysis at the gamma point on the energy-minimized supercell to obtain the complete set of vibrational eigenvectors and eigenvalues [15] [13]. This step diagonalizes the dynamical matrix to identify all vibrational modes, including propagons, diffusons, and locons.
Molecular Dynamics Simulation: Conduct equilibrium MD simulations using appropriate interatomic potentials (e.g., Tersoff for a-Si [13] or ReaxFF for polymers [15]) across the target temperature range. Record atomic positions and velocities at regular intervals to capture the system's dynamical evolution.
Modal Projection: Project the atomic velocities from MD trajectories onto the normal mode eigenvectors obtained in step 2. This critical step decomposes the atomic motion into contributions from each vibrational mode, enabling the calculation of mode-specific heat fluxes [15].
Heat Flux Calculation: Compute the time history of each mode's contribution to the heat current operator using the expression: ( \vec{J}n = \frac{1}{V} \left[ \sumi Ei \vec{v}{i,n} + \frac{1}{2} \sum{i,j} \vec{r}{ij} (\vec{F}{ij} \cdot \vec{v}{j,n}) \right] ), where ( \vec{v}_{i,n} ) is the contribution of mode n to the velocity of atom i [15].
Green-Kubo Integration: Calculate the thermal conductivity contribution for each mode by integrating the autocorrelation of its heat flux: ( \kappa{(n)} = \frac{1}{kB T^2 V} \int0^\infty \langle \vec{J}n(t) \cdot \vec{J}_n(0) \rangle dt )citation:4].
Quantum Correction: Apply a quantum correction to the specific heat to account for quantum effects in mode occupations, particularly important at low temperatures: ( \kappa{(n),Q}(T) = fQ(n,T) \cdot f_\kappa(n,T) )citation:4].
Validation and Benchmarking Procedures
To ensure computational accuracy, GKMA implementations should be validated against established experimental results and benchmarked with other computational approaches. The recent "Phonon Olympics" initiative provides a valuable framework for such validation, bringing together developers and expert users to benchmark open-source thermal conductivity packages [17]. For amorphous silica, direct comparison with experimental thermal conductivity measurements across the 400-800 K temperature range serves as an excellent validation test [15]. Additionally, comparing GKMA predictions for crystalline silicon with results from established methods like the phonon gas model provides further verification of the implementation [15].
Visualization of Thermal Transport Mechanisms
The GKMA framework provides unprecedented visualization of heat conduction mechanisms by quantifying each vibrational mode's contribution to thermal transport. This capability reveals fundamental differences in how heat propagates through ordered versus disordered structures. In crystalline materials, heat transport is dominated by propagating modes (propagons) with well-defined group velocities, while in amorphous systems, significant contributions emerge from non-propagating modes (diffusons and locons) that operate through fundamentally different mechanisms [15] [13].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of GKMA requires both computational tools and specialized material systems for experimental validation. The following toolkit outlines essential components for research in this field:
Table: Essential Research Toolkit for GKMA and Thermal Transport Studies
| Tool Category | Specific Examples | Function/Application | Relevance to GKMA |
|---|---|---|---|
| Computational Codes | ALAMODE, phono3py, ShengBTE [17] | First-principles thermal conductivity calculation | Benchmarking and comparison |
| Interatomic Potentials | Tersoff (a-Si, c-Si), ReaxFF (polymers) [15] [13] | Define atomic interactions in MD simulations | Critical for accurate dynamics |
| Model Systems | a-SiO₂, a-Si, c-Si, Polythiophene [15] [16] [13] | Well-characterized reference materials | Method validation and testing |
| Experimental Validation | Inelastic X-ray Scattering (IXS) [18] | Measures phonon lifetimes and frequencies | Direct experimental comparison |
| Synthesis Methods | Electrolyte gating (La₀.₅Sr₀.₅CoO₃-δ) [19] | Tunes thermal conductivity "on the fly" | Creates materials with tunable properties |
The Green-Kubo Modal Analysis method represents a paradigm shift in thermal transport theory, offering a unified formalism that bridges the historical divide between crystalline and amorphous materials research. By transcending the limitations of the phonon gas model, GKMA provides a comprehensive framework that accurately predicts thermal conductivity across diverse material classes while revealing previously inaccessible mechanisms of heat conduction. The method's validation through experimental studies of amorphous silica and its application to complex systems like polymer chains demonstrates both its robustness and versatility.
Future research directions will likely focus on expanding GKMA's application to increasingly complex material systems, including heterogeneous interfaces, nanocomposites, and biological materials. The integration of machine learning approaches with GKMA, as demonstrated in studies of germanium telluride [18], presents a promising avenue for accelerating calculations and extending the method to more complex potentials. Additionally, the continued development of experimental techniques for measuring thermal properties at extreme conditions [20] will provide crucial validation data across wider parameter spaces. As computational power increases and algorithms refine, GKMA is poised to become an indispensable tool for the rational design of thermal materials with tailored properties for energy applications, electronic cooling, and sustainable technologies.
Expanding the Physical Picture Beyond the Phonon Gas Model
The Phonon Gas Model (PGM) has long been the dominant theoretical framework for interpreting and predicting thermal conductivity in crystalline materials. Its success hinges on treating heat-carrying lattice vibrations (phonons) as a gas of particle-like entities, each with a well-defined mean free path, group velocity, and relaxation time [5]. In ordered, crystalline systems, these properties can be rigorously defined due to the material's periodicity, allowing for highly accurate first-principles calculations of thermal transport [5].
However, this model faces fundamental challenges when applied to disordered solids, such as amorphous silicon dioxide (a-SiO₂) or polymers. The core issue is the lack of long-range periodicity, which makes it impossible to rigorously define a phonon dispersion relation and, consequently, a group velocity for vibrational modes [5]. The PGM becomes highly questionable for the majority of modes in amorphous materials, which are non-propagating diffusons and localized modes (locons), rather than propagating waves [5]. This theoretical gap hinders the accurate prediction and understanding of thermal transport in a vast class of materials, including glasses, amorphous semiconductors, and disordered polymers, which exhibit thermal conductivities that can span three orders of magnitude [21].
Green-Kubo Modal Analysis (GKMA): A Unified Formalism
The Green-Kubo Modal Analysis (GKMA) method has been developed as a unified formalism to directly calculate individual phonon or normal mode contributions to thermal conductivity, overcoming the limitations of the PGM [3]. Its primary advantage is its generality; it can be applied to any type of solid—amorphous materials, crystalline solids, crystalline alloys, polymers, and even molecules—within a single, consistent framework [3].
The GKMA method combines the Green-Kubo (GK) formula, which relates thermal conductivity to the auto-correlation of the heat current in molecular dynamics (MD) simulations, with modal information obtained from lattice dynamics (LD) calculations [5]. This powerful synergy allows GKMA to incorporate all degrees of anharmonicity inherent in the atomic interactions, providing a more complete physical picture than methods like the Allen-Feldman (A-F) theory, which does not fully account for anharmonic effects [5].
Theoretical Formalism
The GKMA formalism begins by computing the normal mode eigenvectors from a lattice dynamics calculation of the entire atomic supercell. The atomic velocities from an MD simulation are then projected onto these normal mode eigenvectors, yielding the time history of each normal mode's amplitude [5]. The critical step is the decomposition of the heat current operator. Each atom's instantaneous velocity is broken down into individual mode contributions, which are then substituted into the heat flux operator. The total heat flux is the sum of all individual mode contributions [5]: [ \vec{J}{total} = \sum{n=1}^{3N} \vec{J}(n) = \frac{1}{V} \left[ \sum{i=1}^{N} Ei \vec{v}i + \frac{1}{2} \sum{i=1}^{N} \sum{j \neq i}^{N} (\vec{F}{ij} \cdot \vec{v}j) \vec{r}{ij} \right] ] where (n) is the mode index, (N) is the total number of atoms, (V) is the supercell volume, (Ei) is the energy of atom (i), (\vec{v}i) is its velocity, (\vec{F}{ij}) is the force between atoms (i) and (j), and (\vec{r}{ij}) is the distance between them [5].
By substituting the modal heat fluxes into the Green-Kubo expression for thermal conductivity, one obtains the thermal conductivity as a direct summation over individual mode contributions, including mode-mode cross-correlations [5]: [ \kappa = \sum{n=1}^{3N} \kappa(n) = \frac{V}{kB T^2} \sum{n=1}^{3N} \int0^{\infty} \langle \vec{J}(n, t) \cdot \vec{J}(n, 0) \rangle dt ] This formulation provides immense insight into the interactions between different modes.
Accounting for Temperature Dependence
A key strength of GKMA is its ability to accurately capture the temperature dependence of thermal conductivity, which is crucial for amorphous materials. This is achieved through a quantum correction function, (fQ), applied to the GKMA-derived mode contributions, (f\kappa) [5]: [ \kappa(T) = \sum{n=1}^{3N} fQ(\omegan, T) \cdot f\kappa(n, T) ] The function (fQ) represents the ratio of quantum to classical specific heat for each mode, which is essential for restoring the proper temperature scaling, especially at low temperatures. The MD-derived (f\kappa) inherently includes the temperature dependence of modal interactions due to anharmonicity [5].
Key Insights from GKMA Application: The Case of Amorphous Silica
Applications of GKMA to materials like amorphous silicon dioxide (a-SiO₂) have yielded critical insights that challenge the conventional PGM-based understanding.
Non-Negligible Role of Localized Modes
A pivotal finding from GKMA is the significant contribution of localized modes (locons) to the total thermal conductivity in a-SiO₂. Previously, these highly localized vibrations were thought to have a negligible contribution to heat transport due to their non-propagating nature [5]. However, GKMA reveals that locons contribute more than 10% to the total thermal conductivity in the temperature range of 400 K to 800 K [5]. Furthermore, these localized modes are largely responsible for the increase in thermal conductivity of a-SiO₂ above room temperature—an effect that neither the PGM nor the A-F model can adequately explain [5].
Quantitative Analysis of Mode Contributions
The table below summarizes the distinct vibrational modes in amorphous materials and their characteristics as revealed by GKMA, providing a quantitative comparison to the traditional PGM view:
Table 1: Modal Contributions to Thermal Conductivity in Amorphous Silicon Dioxide (a-SiO₂) as Revealed by GKMA
| Mode Type | Description | PGM-Based Assumption | GKMA Revelation (400K-800K) |
|---|---|---|---|
| Propagons | Low-frequency, propagating, plane-wave-like vibrations. | Primary heat carriers. | Significant contributors, but not the sole carriers. |
| Diffusons | Medium-frequency, non-propagating, diffusive vibrations. | Contribute to thermal conductivity via diffusion. | Remain the dominant contributors to heat transport. |
| Locons(Localized Modes) | High-frequency, highly localized vibrations. | Negligible contribution to thermal conductivity. | >10% contribution to total κ; key driver of κ increase above room temperature [5]. |
This data underscores a fundamental expansion of the physical picture: thermal transport in disordered materials is not solely governed by propagating waves but involves a complex interplay between propagating, diffusive, and even localized vibrations.
Experimental Protocols and Computational Methodologies
GKMA Workflow for Disordered Solids
The following diagram outlines the core computational workflow for applying GKMA to a disordered solid, illustrating the integration of lattice dynamics and molecular dynamics.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
Implementing the GKMA method requires a combination of robust computational tools and theoretical components. The following table details the essential "research reagents" for this field.
Table 2: Essential Research Reagents and Computational Solutions for GKMA Studies
| Item/Reagent | Function/Description | Application Note |
|---|---|---|
| Interatomic Potential | A classical potential (e.g., SW, Tersoff, ReaxFF) defining atomic interactions. | Potential must accurately describe bonding in disordered systems. GKMA is compatible with multi-body potentials [5]. |
| Molecular Dynamics (MD) Engine | Software (e.g., LAMMPS, GROMACS) to simulate atomic trajectories. | Used to generate the time-history of atomic velocities in NVE/NVT ensembles for heat flux calculation [5]. |
| Lattice Dynamics (LD) Code | Code to perform dynamical matrix diagonalization. | Calculates normal mode eigenvectors and frequencies (at gamma point) for the entire supercell [5]. |
| GKMA Post-Processor | Custom code to project MD velocities onto LD modes and compute modal heat fluxes. | The core implementer of the GKMA formalism, calculating κ(n) via the Green-Kubo formula [5]. |
| Quantum Correction Function (f_Q) | (fQ = \frac{C{q}(\omega, T)}{C_{c}(\omega, T)}), ratio of quantum to classical specific heat. | Critical for obtaining physical results at low temperatures; applied post-processing to MD-derived κ(n) [5]. |
Integration with Modern Data-Driven Methods
A modern extension of this workflow involves integrating generative deep learning with GKMA-accurate methods for high-throughput materials discovery. The following diagram illustrates this unified framework for predicting materials with ultrahigh thermal conductivity, as applied to carbon polymorphs [22].
This unified framework, as demonstrated in a study screening 100,000 carbon candidates, leverages a generative model to create novel structures, which are then distilled using machine-learned interatomic potentials (MLIPs) and structural symmetry metrics [22]. Promising candidates undergo high-fidelity lattice thermal conductivity (κL) evaluation, where protocols like GKMA can be applied. An active learning strategy ("Query by Committee") is often used to selectively improve the MLIPs on the fly, ensuring high-fidelity predictions of stability and thermal properties with minimal computational cost [22]. This entire loop enables the identification of novel materials, such as carbon polymorphs with ultrahigh thermal conductivity, validated against established data [22].
The Green-Kubo Modal Analysis method provides a powerful and unified formalism for expanding the physical picture of thermal transport beyond the confines of the Phonon Gas Model. By enabling the direct calculation of individual mode contributions to thermal conductivity in both ordered and disordered materials, GKMA has uncovered previously overlooked mechanisms, such as the significant role of localized modes in amorphous silica. The insights provided by GKMA, combined with emerging data-driven methods, are paving the way for a more profound and general understanding of heat conduction in materials, accelerating the discovery of new materials with tailored thermal properties for energy conversion and management technologies.
Implementing GKMA: From Theory to Molecular Dynamics Practice
Decomposing Thermal Conductivity into Modal Contributions
Understanding thermal transport in disordered solids—such as amorphous materials, polymers, and crystalline alloys—is a fundamental challenge in materials science and thermal engineering. The Green-Kubo Modal Analysis (GKMA) method represents a significant theoretical and computational advancement by enabling the direct calculation of individual phonon or normal mode contributions to the overall thermal conductivity within a unified formalism [3]. This Application Note provides a detailed framework for applying GKMA to decompose thermal conductivity in disordered systems, a capability critical for optimizing materials for applications ranging from thermal energy grid storage to advanced semiconductor packaging [23] [3].
Theoretical Background: The GKMA Formalism
The Green-Kubo method relates the macroscopic thermal conductivity (κ) to the microscopic behavior of the atomic system, specifically the time evolution of the heat current. For a system in equilibrium, the thermal conductivity tensor is given by the integral of the heat current autocorrelation function (HCACF) [24]:
$$κ{αβ} = \frac{1}{V kB T^2} \int0^∞ \left< Jα(0) J_β(t) \right> dt$$
where:
- (V) is the system volume
- (k_B) is Boltzmann's constant
- (T) is the absolute temperature
- (J_α(t)) is the α-component of the heat current vector at time (t$
- ( \left< ... \right> ) denotes an ensemble average
The GKMA formalism's key innovation is its ability to decompose the total heat current, (J{total}), into contributions from individual normal modes or phonons: (J{total} = \sum{λ} J{λ}). This decomposition allows the total HCACF to be expressed as a sum of auto-correlation and cross-correlation terms between these modal contributions [3]. Consequently, the thermal conductivity can be partitioned as:
$$κ{total} = \sum{λ} κ{λ}^{auto} + \sum{λ \neq μ} κ_{λ, μ}^{cross}$$
where (κ{λ}^{auto}) is the contribution from mode (λ), and (κ{λ, μ}^{cross}) represents the energy transfer between modes (λ) and (μ). This decomposition is pivotal for identifying the specific modes that dominate heat transport in complex, disordered solids, where the traditional phonon gas model (PGM) often provides an incomplete physical picture [3].
Experimental Protocols and Workflows
Protocol 1: GKMA Workflow for Disordered Solids
Objective: To computationally determine the modal decomposition of thermal conductivity for a disordered solid (e.g., an amorphous polymer or a Metal-Organic Framework) using GKMA.
Step 1: System Preparation
- Model Construction: Generate an atomistic model of the disordered solid. For amorphous systems, this typically involves melt-quenching using classical molecular dynamics (MD) to create a structurally realistic sample.
- Energy Minimization: Relax the generated structure using a conjugate gradient or steepest descent algorithm to eliminate unrealistically high atomic forces.
- Equilibration: Perform NPT (constant Number of particles, Pressure, and Temperature) MD simulation to equilibrate the system density at the target temperature and pressure.
Step 2: Normal Mode Calculation
- Snapshot Selection: Extract multiple statistically independent snapshots from the equilibrated MD trajectory.
- Hessian Matrix: For each snapshot, compute the mass-weighted Hessian (force constant) matrix ( \frac{1}{\sqrt{mi mj}} \frac{\partial^2 E}{\partial ri \partial rj} ) where (E) is the potential energy and (r_i) is the position of atom (i$.
- Diagonalization: Diagonalize the Hessian matrix to obtain the eigenvalues (squared frequencies, (ω_λ^2)) and eigenvectors (polarization vectors) for each normal mode (λ).
Step 3: Equilibrium Molecular Dynamics (EMD) Simulation
- Simulation Run: Conduct a microcanonical (NVE) MD simulation starting from an equilibrated configuration.
- Data Harvesting: Output the atomic positions, velocities, and stresses at a high frequency (e.g., every 1-10 fs) to ensure accurate resolution of the heat current fluctuations.
Step 4: Heat Current Decomposition and HCACF Truncation
- Modal Heat Currents: Using the saved MD trajectory and the normal mode eigenvectors, compute the instantaneous heat current contribution (J_λ(t)) for each mode of interest at each time step [3].
- HCACF Calculation: Compute the total and modal HCACFs, ( \left< J{total}(0) J{total}(t) \right> ) and ( \left< J{λ}(0) J{λ}(t) \right> ).
- Truncation Time: A critical step is determining the appropriate truncation time for the HCACF integral. Follow the method from recent studies: analyze the decay of the HCACF and identify the time at which it converges to zero or becomes statistically noisy. For MOFs, a systematic method for this truncation has been devised to ensure reliability [24].
Step 5: Thermal Conductivity Integration
- Numerical Integration: Numerically integrate the truncated HCACF for the total and modal contributions according to the Green-Kubo formula.
- Ensemble Averaging: Average the results over multiple independent simulation runs starting from different initial conditions to obtain a statistically robust value for (κ{total}) and the decomposed (κ{λ}).
The following workflow diagram illustrates this comprehensive protocol:
Protocol 2: Validation Against Experimental Data
Objective: To validate the GKMA-predicted thermal conductivity values against experimental measurements.
Step 1: Experimental Measurement
- Sample Preparation: Synthesize or procure a bulk sample of the material under study.
- Standardized Testing: Measure the bulk thermal conductivity using a standardized method such as the transient plane source (TPS) technique or laser flash analysis. Ensure measurement conditions (temperature, pressure) match the simulation conditions.
Step 2: Data Comparison and Analysis
- Direct Comparison: Compare the experimentally measured thermal conductivity value with the GKMA-calculated (κ_{total}).
- Uncertainty Quantification: Calculate the percentage difference and ensure it falls within acceptable bounds (e.g., <10-15% for many solids). As noted in GKMA research, excellent agreement with experimental data has been observed, though continued testing is warranted [3].
The Scientist's Toolkit: Research Reagent Solutions
Table 1: Essential Materials and Computational Tools for GKMA Studies
| Item Name | Function/Description | Application Context in GKMA |
|---|---|---|
| Molecular Dynamics Software (e.g., LAMMPS, GROMACS) | Open-source or commercial software packages to perform the energy minimization, equilibration, and production EMD simulations. | Core platform for generating the atomic trajectory data required for the Green-Kubo analysis [24]. |
| Interatomic Potentials (Force Fields) | A set of mathematical functions and parameters describing the potential energy surface and forces between atoms. | Critical for simulating realistic atomic interactions. Accuracy directly impacts the reliability of the heat current and HCACF. |
| Normal Mode Analysis Code | Custom or integrated code to compute the Hessian matrix and diagonalize it to obtain eigenfrequencies and eigenvectors. | Enables the decomposition of the atomic dynamics into individual modes, which is the foundation of the "Modal Analysis" [3]. |
| Post-processing Scripts | Custom scripts (e.g., in Python) to compute the heat current from MD trajectories, perform the decomposition, and integrate the HCACF. | Essential for implementing the GKMA formalism, linking raw simulation data to the final thermal conductivity values. |
| High-Performance Computing (HPC) Cluster | A computing system with many processors and large memory, capable of handling thousands of atoms for nanosecond-scale simulations. | GKMA is computationally demanding; EMD and Hessian diagonalization for large, disordered systems require significant HPC resources. |
Data Presentation and Analysis
The following tables summarize key quantitative findings and parameters relevant to thermal conductivity analysis and GKMA applications.
Table 2: Projected Global Market for Thermal Conductive Materials (2021-2033)This market data reflects the growing industrial application and importance of advanced thermal management materials, underscoring the relevance of fundamental research into thermal conductivity. [25]
| Region | Market Size 2021 (USD Million) | Projected Market Size 2025 (USD Million) | Projected Market Size 2033 (USD Million) | CAGR (2025-2033) |
|---|---|---|---|---|
| Global | 920.1 | 1310.4 | 2658 | 9.24% |
| North America | 238.3 | 329.2 | 647.0 | 8.81% |
| Europe | 200.0 | 287.6 | 589.3 | 9.38% |
| Asia Pacific | 203.2 | 288.0 | 581.3 | 9.18% |
Table 3: Benchmarking Common Thermal Interface Material (TIM) FillersThe properties of fillers influence the macroscopic thermal conductivity of composite materials. Understanding single-component conductivity is a stepping stone to analyzing complex solids. [23]
| Filler Material | Typical Thermal Conductivity (W/m·K) | Key Properties and Notes |
|---|---|---|
| Alumina (Al₂O₃) | ~30 | Low cost, electrically insulating, widely used. |
| Boron Nitride (BN) | ~50-600 (anisotropic) | Electrically insulating, high thermal conductivity, often used in sheet form. |
| Aluminum Nitride (AlN) | ~150-200 | High conductivity, good electrical insulation, but higher cost. |
| Silver (Ag) | ~429 | Highest electrical and thermal conductivity, but expensive and electrically conductive. |
| Graphene | ~2000-5000 | Extremely high in-plane thermal conductivity, subject of intense research for high-performance applications [23]. |
Discussion and Outlook
The GKMA method provides a powerful lens through which to view thermal transport in disordered solids. Initial applications have already revealed that the conventional phonon gas model does not fully capture the physical picture, indicating the presence of modes with behaviors outside this model's scope [3]. Future research directions will likely focus on:
- Expanding the Physical Picture: Developing a more general model of thermal transport that can account for the unique behaviors of modes in disordered systems identified by GKMA.
- Application to Novel Materials: Utilizing GKMA to screen and design new materials for specific thermal applications, such as thermal energy grid storage (TEGS) or thermal interface materials for EV power electronics and data centers, where managing higher power densities and junction temperatures is critical [23] [3].
- Methodological Refinements: Continued improvement of protocols, particularly for robust and automated HCACF truncation in increasingly complex material systems like Metal-Organic Frameworks (MOFs) [24].
The ability to decompose thermal conductivity into its fundamental modal contributions is not merely an academic exercise; it is a critical tool for the rational design of next-generation materials needed to solve pressing challenges in energy storage, electronics cooling, and sustainable technology.
Integrating Lattice Dynamics with the Green-Kubo Formula
The Green-Kubo (GK) formalism, which relates the thermal conductivity of a material to the time integral of the heat current autocorrelation function, is a cornerstone of equilibrium molecular dynamics (EMD) simulations [26]. For disordered solids such as amorphous materials and polymers, the conventional phonon gas model (PGM) faces significant challenges as it relies on well-defined phonon velocities, a concept difficult to establish in non-crystalline systems [3] [27].
Green-Kubo Modal Analysis (GKMA) addresses this limitation by providing a unified formalism that integrates lattice dynamics with the Green-Kubo formula. This method enables the direct calculation of individual phonon or normal mode contributions to thermal conductivity, offering a generalizable approach applicable to amorphous materials, crystalline solids, alloys, and polymers [3] [27]. This Application Note details the protocols for implementing GKMA, specifically tailored for investigating thermal transport in disordered solids.
Theoretical Foundation
The foundational Green-Kubo expression for calculating lattice thermal conductivity (( \kappa )) is:
$$ \kappa = \frac{V}{kB T^2} \int0^\infty \langle J(t) \cdot J(0) \rangle dt $$
where:
- ( V ) is the system volume
- ( k_B ) is the Boltzmann constant
- ( T ) is the temperature
- ( J ) is the microscopic heat current vector
- The angle brackets denote an ensemble average [26] [28]
The GKMA method extends this foundation by decomposing the total heat current into contributions from specific atomic interactions or normal modes, thereby providing a modal analysis of thermal transport [3]. The total heat flux ( J(t) ) in an atomic system can be decomposed into two primary components [28]:
- Convective term (( J_c )): Arises from the diffusion of particles and their associated energies.
- Virial term (( J_v )): Arises from atomic interactions through interatomic forces.
This decomposition allows for a detailed analysis of the correlation functions that constitute the thermal conductivity:
$$ \kappa = \kappa{cc} + \kappa{vv} + \kappa{cv} + \kappa{vc} $$
where the subscripts denote the auto-correlation (( \kappa{cc}, \kappa{vv} )) and cross-correlation (( \kappa{cv}, \kappa{vc} )) contributions of the convective and virial heat flux components [28].
Table 1: Key Formulas in Green-Kubo Analysis for Thermal Conductivity
| Formula Name | Mathematical Expression | Application Context |
|---|---|---|
| Standard Green-Kubo | ( \kappa = \frac{V}{kB T^2} \int0^\infty \langle J(t) \cdot J(0) \rangle dt ) | General purpose EMD; total ( \kappa ) calculation [26] [28] |
| Multi-Component PC Formula | ( \kappa2 = \frac{1}{T^2}\left( L{QQ} - \frac{L{Q1}L{1Q}}{L_{11}} \right) ) | Binary systems with atomic diffusion; more accurate [26] |
| Virial Heat Flux | ( Jv = \frac{1}{2V} \left[ \sum{i |
Atomistic interaction contribution to heat flow [28] |
Computational Setup and Protocols
Software and Forcefields
Primary MD Engine: The LAMMPS (Large-scale Atomic/Molecular Massively Parallel Simulator) code is universally employed for performing the molecular dynamics simulations required for GKMA [26] [28]. Its stability and built-in functions for calculating heat flux are essential.
Forcefield Selection: The choice of interatomic potential is critical. The search results highlight:
- Buckingham potentials for ionic materials like Li₂O and TiO₂, combined with Ewald summation for long-range Coulombic interactions [26].
- OPLS-AA (Optimized Potentials for Liquid Simulations - All Atom) for organic molecules like alcohols, which explicitly defines bonded and non-bonded interactions [28].
System Preparation and Simulation Parameters
Table 2: Representative Simulation Parameters from Literature
| Parameter | Crystalline/Ionic Solids (e.g., Li₂O, TiO₂) [26] | Molecular Liquids (e.g., Alcohols) [28] | Disordered Solids (e.g., a-Si, a-SiO₂) [27] |
|---|---|---|---|
| Ensemble | NPT (temperature & pressure control) | NPT | NPT or NVT (constant volume) |
| Temperature Control | Nose-Hoover thermostat | Nose-Hoover thermostat | Nose-Hoover thermostat |
| Time Step | 1.0 fs | Typically 1-2 fs | 1.0 fs |
| Total Simulation Time | ~ 1 ns (for production) | Sufficient for correlation decay | Requires convergence testing |
| System Size | 1000+ atoms | 100s of molecules | 1000+ atoms to model disorder |
GKMA Workflow and Signaling Pathways
The following workflow diagram outlines the core procedural sequence for applying GKMA, from initial configuration to final analysis.
Step-by-Step Protocol
System Preparation
- For crystals: Build a supercell from the crystallographic unit cell.
- For disordered solids (a-Si, a-SiO₂): Use a melt-and-quench procedure or experimental structure factors to generate an atomic configuration that accurately represents the amorphous phase [27].
- For molecules: Pack multiple molecules into a simulation box at the experimental density.
Equilibration Phase
- Run an NPT simulation to relax the system to the target temperature and pressure, ensuring correct density.
- This is followed by an NVE (microcanonical) simulation to ensure the system is in a stable equilibrium state before production runs.
Production Phase
- A lengthy NVE simulation is performed. During this run, the atomic positions, velocities, and stresses are sampled at frequent intervals (e.g., every 10-100 time steps).
- This trajectory data is used for subsequent heat flux calculation.
Heat Flux Decomposition
- The total heat current ( J(t) ) is calculated for each saved timestep using the virial and convective formulations [28].
- For GKMA: The key step is decomposing this total heat flux into contributions from specific normal modes or atomic pairs. This is the core of the modal analysis, moving beyond the total correlation ( \langle J(t) \cdot J(0) \rangle ) to correlations between specific modal currents ( \langle J\mu(t) \cdot J\nu(0) \rangle ) [3].
Correlation Function Calculation
- The auto-correlation function of the total heat flux, or the cross-correlation functions of the decomposed modal fluxes, are calculated.
- The correlation function is integrated over time to obtain the running thermal conductivity integral. The value is taken as the average over the plateau region of this integral.
Data Analysis and Validation
Analyzing Correlation Components
The decomposed heat flux allows for the calculation of distinct thermal conductivity components, providing insight into the mechanisms of heat transfer [28]:
- ( \kappa_{vv} ): The virial auto-correlation, representing heat conduction through direct atomic interactions.
- ( \kappa_{cc} ): The convective auto-correlation, representing heat transport via atomic diffusion.
- ( \kappa{cv}, \kappa{vc} ): The cross-correlations, indicating collaborative or competitive relationships between different transport mechanisms.
Atomic-Level Pairwise Breakdown
For molecular systems, the virial component ( Jv ) can be broken down further into contributions from specific atomic pairs (e.g., C-C, O-H, C-O) [28]. This pairwise breakdown, expressed as ( Jp = \sum J_{XY} ), allows for the calculation of:
- Auto-correlations (( \lambda_{XX-XX} )): The contribution from a single type of atomic interaction.
- Cross-correlations (( \lambda_{XX-YY} )): The collaborative (positive) or competitive (negative) interplay between different atomic interactions.
The following diagram illustrates the hierarchical decomposition of thermal conductivity within the GKMA framework, from the total value down to the atomic pairwise contributions.
Validation and Error Checking
- Convergence Test: The simulation must be run long enough for the Green-Kubo integral to reach a stable plateau. The required correlation time can be system-dependent.
- Formula Selection: As highlighted in the study of binary systems (e.g., Li₂O, TiO₂), the choice of Green-Kubo formula is critical when significant atomic diffusion is present. The formula ( \kappa2 ), which accounts for cross-phenomenological coefficients, is more accurate than the simpler ( \kappa1 ) in such cases [26].
- Experimental Comparison: Validate simulation results against experimental data for similar materials where available, acknowledging that simulations typically calculate only the lattice contribution.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools and Materials for GKMA Simulations
| Item Name | Function/Description | Example Use Case |
|---|---|---|
| LAMMPS MD Package | Primary simulation engine for performing equilibrium MD and calculating heat flux. | Core software for all simulation steps [26] [28]. |
| Buckingham Potential | An interatomic potential model combining exponential repulsion with Coulomb and dispersion terms. | Simulating ionic solids like Li₂O and oxides like TiO₂ [26]. |
| OPLS-AA Forcefield | An all-atom forcefield for organic molecules defining bonds, angles, dihedrals, and non-bonded interactions. | Simulating molecular systems like alcohols [28]. |
| Ewald Summation | A method for handling long-range Coulombic interactions in periodic systems. | Essential for simulating ionic materials [26]. |
| Nose-Hoover Thermostat | A deterministic algorithm for maintaining constant temperature (NVT) or pressure (NPT) during MD. | Standard for equilibration and production runs [26] [28]. |
Calculating Mode-Wise Heat Current from MD Trajectories
The Green-Kubo Modal Analysis (GKMA) method represents a significant advancement for researchers investigating thermal transport in disordered solids, such as amorphous materials. This technique connects macroscopic thermal conductivity to the microscopic energy carriers within a material. For disordered solids, where traditional particle-like phonon models often fail due to the ill-defined nature of vibrational modes, GKMA provides a more robust framework for understanding thermal conductivity [13]. It enables the decomposition of the overall heat flux into contributions from specific atomic interactions or vibrational modes, offering unprecedented insight into the nanoscale mechanisms of thermal conduction. This protocol details the application of GKMA for calculating mode-wise heat current from molecular dynamics (MD) trajectories, with specific considerations for the analysis of disordered solids.
Theoretical Foundation
The Green-Kubo Formalism
The Green-Kubo method calculates thermal conductivity (( \lambda )) by integrating the heat current autocorrelation function (HCACF) over time, as derived from fluctuation-dissipation theory [12]. The fundamental equation is:
[ \lambda = \frac{V}{kB T^2} \int0^{\infty} \langle \mathbf{J}(t) \cdot \mathbf{J}(0) \rangle dt ]
Here, (V) is the system volume, (T) is temperature, (k_B) is Boltzmann's constant, and (\mathbf{J}(t)) is the heat flux vector at time (t). The angle brackets (\langle \cdots \rangle) denote an ensemble average over an equilibrium molecular dynamics trajectory [12] [29].
Heat Current Formulation
The total heat current (\mathbf{J}(t)) is a sum of convective and virial components [12]:
[ \mathbf{J}(t) = \mathbf{J}c(t) + \mathbf{J}v(t) ]
The convective term ((\mathbf{J}_c)) arises primarily from particle diffusion:
[ \mathbf{J}c = \frac{1}{V} \sumi ei \mathbf{v}i ]
where (ei) and (\mathbf{v}i) are the per-atom energy and velocity of atom (i), respectively.
The virial term ((\mathbf{J}_v)) originates from atomic interactions:
[
\mathbf{J}v = \frac{1}{2V} \sum{i
where (\mathbf{F}{ij}) is the force between atoms (i) and (j), and (\mathbf{r}{ij}) is their separation vector [12].
This decomposition enables the breakdown of thermal conductivity into auto-correlation and cross-correlation components:
[ \lambda = \lambda{cc} + \lambda{vv} + \lambda{cv} + \lambda{vc} ]
where (\lambda{cc}) and (\lambda{vv}) are auto-correlations of convective and virial terms, respectively, and (\lambda{cv}), (\lambda{vc}) are their cross-correlations [12].
Challenges in Disordered Solids
In amorphous materials, the Phonon Gas Model (PGM) faces significant limitations. Most vibrational modes in disordered materials do not propagate, making it difficult to define phonon velocities rigorously due to the lack of periodicity [13]. The GKMA approach addresses this challenge by focusing on the actual energy transport captured in MD simulations without relying on questionable assumptions about phonon propagation.
Table 1: Key Differences Between Crystalline and Disordered Solids for Thermal Analysis
| Aspect | Crystalline Solids | Disordered Solids (Amorphous) |
|---|---|---|
| Periodicity | Long-range order present | No long-range periodicity |
| Phonon Velocity | Well-defined from dispersion | Ill-defined for many modes |
| Dominant Heat Carriers | Propagating vibrations | Non-propagating, diffusive modes |
| GKMA Application | Standard decomposition | Essential for meaningful analysis |
Computational Setup
Research Reagent Solutions
Table 2: Essential Computational Tools for GKMA Implementation
| Tool Category | Specific Implementation | Function/Purpose |
|---|---|---|
| MD Engine | LAMMPS [12] [29] | Primary simulation platform with GKMA capabilities |
| Force Field | OPLS-AA [12] | Alcohols, organic molecules |
| Force Field | Tersoff potential [13] [30] | Covalent materials (Si, Ge, C) |
| Force Field | SB potential [29] | Energetic materials (HMX) |
| Analysis Code | Custom modifications [12] | Atomic-level flux decomposition |
| Long-Range Electrostatics | PPPM method [12] | Coulomb interactions with cutoff |
Simulation Parameters
For accurate thermal conductivity calculations, these parameters should be carefully considered:
- Cutoff Distance: 10-12 Å for short-range interactions [12]
- Long-Range Coulombics: Particle-Particle-Particle-Mesh (PPPM) method with similar cutoff [12]
- Constraint Handling: SHAKE algorithm for bonds involving hydrogen [12]
- Ensemble: NVE for production runs after proper equilibration in NPT ensemble [12]
- Time Step: Typically 0.5-1.0 fs for atomic-level accuracy
GKMA Workflow Implementation
The following diagram illustrates the complete workflow for calculating mode-wise heat current using GKMA:
System Preparation and Equilibration
- Build Amorphous Structure: Create a disordered system using appropriate modeling tools
- Energy Minimization: Reduce high-energy contacts using conjugate gradient or steepest descent algorithms
- NPT Equilibration: Equilibrate density at target temperature and pressure (typically 300 K, 1 atm)
- NVE Equilibration: Switch to microcanonical ensemble for production dynamics
Heat Current Calculation Protocol
The virial component of the heat current can be decomposed into specific atomic interactions, which is particularly valuable for understanding contributions from different chemical groups in complex materials [12]:
[ \mathbf{J}v = \underbrace{\mathbf{J}{ep} + \mathbf{J}{ip} + \mathbf{J}{lc}}{\text{non-bonded}} + \overbrace{\mathbf{J}{b}}^{\text{bonded}} ]
where:
- (\mathbf{J}_{ep}): External pairwise interactions (between molecules)
- (\mathbf{J}_{ip}): Internal pairwise interactions (within molecules)
- (\mathbf{J}_{lc}): Long-range Coulomb interactions
- (\mathbf{J}_{b}): Bonded interactions (bonds, angles, dihedrals)
For atomic-level analysis, pairwise flux can be further decomposed:
[ \mathbf{J}p = \sum \mathbf{J}{XY}, \quad X,Y = C,O,H,\ldots ]
where (\mathbf{J}_{XY}) represents heat flux arising from interactions between atom types X and Y [12].
Mode-Wise Decomposition Algorithm
- Trajectory Processing: Extract atomic positions, velocities, and forces from production MD run
- Heat Flux Calculation: Compute total heat flux using the combined convective and virial formulations
- Atomic Partitioning: Decompose virial heat flux into atomic-level contributions
- Interaction Classification: Categorize contributions by interaction type (C-C, O-H, etc.) and bonding environment
- Correlation Calculation: Compute auto-correlation and cross-correlation functions for each component
Table 3: Correlation Component Interpretation Guide
| Correlation Type | Mathematical Form | Physical Interpretation |
|---|---|---|
| Auto-correlation | (\langle J{XX} \cdot J{XX} \rangle) | Direct contribution from X-X interactions |
| Positive Cross-correlation | (\langle J{XX} \cdot J{YY} \rangle > 0) | Collaborative heat transfer between X and Y |
| Negative Cross-correlation | (\langle J{XX} \cdot J{YY} \rangle < 0) | Competitive relationship between X and Y |
| Zero Cross-correlation | (\langle J{XX} \cdot J{YY} \rangle \approx 0) | Independent heat transfer channels |
Advanced Technical Considerations
Heat Current Filtering
For polyatomic crystals and complex solids, the raw heat current correlation functions often exhibit large-amplitude oscillations that hinder convergence [29]. Filtering out non-contributing components significantly improves results:
- Identify Bounded Functions: Components corresponding to time derivatives of bounded linear functions of atomic positions do not contribute to thermal conductivity
- Apply Fourier Filtering: Remove high-frequency oscillations while preserving physical contributions
- Validate Filtering: Ensure filtered correlation function maintains proper long-time behavior
Convergence Optimization
- Extended Sampling: Run multiple independent trajectories (typically 5-10) with different initial velocities
- Long Time Correlation: Ensure simulation length exceeds correlation time by at least an order of magnitude
- Block Averaging: Use block averaging techniques to estimate statistical uncertainty
- Error Analysis: Calculate standard error across independent trajectories
Disordered Solids Specifics
For amorphous materials, these specific considerations apply:
- Larger System Sizes: Disordered systems require larger simulation cells to capture structural diversity
- Multiple Configurations: Generate and average over multiple independent amorphous structures
- Anharmonic Effects: Properly account for strong anharmonicity in force constants
- Propagating vs. Diffusive Modes: Recognize that most modes in disordered solids are non-propagating [13]
Data Analysis and Interpretation
Thermal Conductivity Calculation
The time-dependent thermal conductivity is computed as:
[ \lambda(t) = \frac{V}{kB T^2} \int0^t \langle \mathbf{J}(0) \cdot \mathbf{J}(\tau) \rangle d\tau ]
The converged value of (\lambda(t)) as (t \to \infty) gives the thermal conductivity. For mode-wise decomposition, this calculation is performed for each correlation component.
Case Study: Alcohol Systems
Applying atomic-level breakdown to small alcohols reveals insightful patterns [12]:
- Hydroxyl groups dominate thermal transport in ethylene glycol
- Cross-correlations between carbon and hydroxyl atoms can be collaborative or competitive
- Adding a carbon atom to ethanol (making propanol) causes minimal conductivity change
- Adding a hydroxyl group to ethanol (making ethylene glycol) significantly increases conductivity
Troubleshooting Common Issues
- Poor Convergence: Extend simulation time, increase system size, implement filtering
- Unphysical Oscillations: Apply current filtering, check force field implementation
- Negative Contributions: Verify these are genuine competitive effects, not numerical artifacts
- Size Dependence: Perform finite-size scaling analysis for accurate bulk values
The GKMA method for calculating mode-wise heat current from MD trajectories provides a powerful framework for understanding thermal transport in disordered solids. By decomposing the heat flux into atomic-level contributions, researchers can identify the specific molecular interactions responsible for heat conduction, enabling targeted molecular design for thermal management applications. For amorphous materials, where traditional phonon models fail, this approach offers a physically rigorous alternative that directly computes thermal conductivity from fundamental atomic interactions.
Step-by-Step Workflow for GKMA Simulation Setup
Green-Kubo Modal Analysis (GKMA) is an advanced computational method for determining individual phonon mode contributions to thermal conductivity in various materials. Unlike the traditional phonon gas model (PGM), which becomes questionable for disordered materials where phonon velocities cannot be rigorously defined, GKMA provides a unified formalism applicable to both crystalline and amorphous solids [15]. This methodology is particularly valuable for studying thermally insulating disordered solids such as amorphous silicon dioxide (a-SiO₂) and amorphous carbon (a-C), where conventional approaches struggle to accurately predict thermal transport properties [10] [15].
The fundamental principle of GKMA involves projecting atomic velocities from molecular dynamics (MD) simulations onto normal mode eigenvectors obtained from lattice dynamics calculations. This projection enables the decomposition of the heat flux operator into individual mode contributions, which are then substituted into the Green-Kubo formula to compute mode-level thermal conductivity [10]. This approach incorporates all anharmonic effects naturally through the MD simulations, providing a more complete picture of thermal transport in complex materials where anharmonicity plays a significant role.
Theoretical Foundation of GKMA
Mathematical Formulation
The GKMA method calculates thermal conductivity using the Green-Kubo formula, which relates the thermal conductivity tensor to the autocorrelation of the heat flux. For a system with N vibrational modes, the thermal conductivity contribution of mode n is given by:
$$\kappa(n) = \frac{V}{kB T^2} \int0^\infty \langle \mathbf{Q}(n,t) \cdot \mathbf{Q}(n,0) \rangle dt$$
where V is the system volume, k_B is Boltzmann's constant, T is temperature, and Q(n,t) is the heat flux vector of mode n at time t [10]. The total thermal conductivity is obtained by summing contributions from all individual modes.
The heat flux operator for mode n is derived by substituting the modal components of atomic velocities into the Hardy expression for heat flux:
$$\mathbf{Q}(t) = \sumn \mathbf{Q}(n,t) = \sumn \left[ \sumi \frac{d\mathbf{r}i}{dt} Ei - \sum{i,j>i} \frac{d\mathbf{r}i}{dt} \cdot \frac{\partial \Phi}{\partial \mathbf{r}{ij}} \mathbf{r}_{ij} \right]^{(n)}$$
where the superscript (n) indicates the portion of the quantity associated with mode n [15].
Phase Quotient Analysis
For disordered materials, the Phase Quotient (PQ) provides a means to characterize vibrational modes as acoustic-like or optical-like based on their atomic displacement patterns. The PQ for mode n is defined as:
$$PQ(n) = \frac{\summ \vec{e}i(n) \cdot \vec{e}j(n)}{\summ |\vec{e}i(n) \cdot \vec{e}j(n)|}$$
where atoms i and j constitute the m-th bond, and e_i(n) is the eigenvector of atom i for mode n [10]. Modes with positive PQ values exhibit acoustic-like character (in-phase atomic motions), while those with negative PQ display optical-like character (out-of-phase atomic motions). This distinction is particularly important in disordered solids where traditional acoustic/optical classifications break down.
Computational Setup and Workflow
Software and Hardware Requirements
Table 1: Essential Software Components for GKMA Simulations
| Software Tool | Purpose | Key Features |
|---|---|---|
| GPUMD | Molecular dynamics simulations | Optimized for GPU computing, implements GKMA and HNEMA methods [31] [32] |
| gpyumd | Python interface for GPUMD | Processes inputs/outputs, facilitates simulation setup and data analysis [31] |
| ASE | Atomic simulation environment | Structure creation, manipulation, and analysis [31] |
| VASP/DFT | First-principles calculations | Potential parameterization (optional) |
The GKMA workflow requires significant computational resources, particularly for the molecular dynamics simulations. GPU-accelerated computing is highly recommended through the GPUMD package to reduce computation time for systems with thousands of atoms [31].
Research Reagent Solutions
Table 2: Essential Materials and Computational Components
| Component | Function | Example Application |
|---|---|---|
| Interatomic Potentials | Defines atomic interactions | Tersoff potential for hBN [31]; ReaxFF for polymers [15] |
| Model Structures | Represents material system | amorphous SiO₂, amorphous carbon, InGaAs alloys [10] [15] |
| Supercell Models | Finite representation of materials | ~10×10 nm² hBN sheet with 3840 atoms [31] |
| Quantum Correction | Accounts for quantum effects | Quantum-to-classical specific heat ratio [15] |
Step-by-Step Implementation Protocol
Structure Creation and Preparation
The initial phase involves creating an atomistic model of the material system:
Generate Atomic Structure: Create a representative supercell of the material using ASE or similar tools. For example, a hexagonal boron nitride (hBN) sheet can be modeled using a 3840-atom structure approximately 10×10 nm² in dimension [31]:
Define Basis and k-Points: For modal analysis, each atom requires a unique basis ID, and only the Γ-point (k = [0,0,0]) is needed since the entire supercell is treated as the unit cell [31]:
Eigenvector Calculation
The eigenvectors form the basis for projecting MD trajectories onto normal modes:
Energy Minimization: Relax the structure to eliminate high-energy configurations:
Phonon Calculation: Compute the harmonic force constants and eigenvectors:
The
run.infile for this stage should contain:Prepare Eigenvector File: After the phonon calculation, rename the output file
eigenvector.outtoeigenvector.infor use in subsequent GKMA simulations [31].
Molecular Dynamics Simulation
With the eigenvectors prepared, conduct MD simulations to capture anharmonic effects:
Set Initial Conditions: Define initial velocities corresponding to the target temperature:
Configure Ensemble and Parameters: Set up the MD ensemble and time step:
Enable GKMA Calculation: Activate the modal heat current computation:
Run Production Simulation: Execute the MD simulation for sufficient duration to converge heat flux autocorrelations:
Data Analysis and Thermal Conductivity Calculation
The final stage involves processing the simulation data to obtain thermal conductivity values:
Load Frequency Information: Analyze the vibrational modes and their properties:
Compute Spectral Thermal Conductivity: Calculate the mode-resolved thermal conductivity contributions:
Apply Quantum Correction: Adjust for quantum effects in specific heat:
The quantum correction factor f_Q represents the ratio of quantum to classical specific heat for each mode and is essential for obtaining physically accurate results, particularly at lower temperatures [15].
Application to Disordered Solids
GKMA has revealed several important insights into thermal transport in disordered solids:
- In amorphous SiO₂, localized modes (locons) contribute more than 10% to the total thermal conductivity between 400-800 K, contradicting previous assumptions that locons have negligible contribution [15].
- The thermal conductivity of amorphous SiO₂ increases with temperature above room temperature, an effect that can be explained by the significant contribution of locons revealed through GKMA [15].
- In disordered materials, modes with negative phase quotient (optical-like character) make non-negligible contributions to thermal transport, unlike in crystalline materials where optical phonon contributions are typically small [10].
Table 3: GKMA Applications to Representative Disordered Materials
| Material | Key Finding | Temperature Range | Computational Requirements |
|---|---|---|---|
| a-SiO₂ | Locons contribute >10% to κ above 400 K | 400-800 K | ~1000 atoms, 1M+ MD steps [15] |
| a-C | Significant negative-PQ mode contributions | 300-600 K | ~1000 atoms, 1M+ MD steps [10] |
| InGaAs Alloy | Redefined acoustic/optical demarcations | 300 K | Supercell with disorder [10] |
Troubleshooting and Best Practices
Successful implementation of GKMA simulations requires attention to several critical factors:
System Size Convergence: Ensure the supercell is sufficiently large to capture relevant phonon modes. For disordered systems, typically 1000-4000 atoms are necessary to obtain representative results [31] [15].
Statistical Sampling: Run multiple independent simulations with different initial random seeds to improve statistical accuracy, particularly for the heat flux autocorrelation function.
Quantum Corrections: Always apply quantum corrections to specific heat when comparing with experimental data, especially at temperatures below the Debye temperature [15].
Potential Validation: Verify that the interatomic potential accurately reproduces material structure and vibrational properties before proceeding with GKMA calculations.
Convergence Testing: Monitor the convergence of cumulative thermal conductivity with respect to both correlation time and simulation duration to ensure adequate sampling.
This comprehensive workflow provides researchers with a robust protocol for implementing GKMA simulations to investigate thermal transport in disordered solids, enabling insights that bridge the gap between crystalline and amorphous materials within a unified theoretical framework.
The Green-Kubo Modal Analysis (GKMA) method represents a significant advancement in computational materials science, providing a unified formalism to calculate modal contributions to thermal conductivity (κ) in both crystalline and amorphous materials. Unlike the traditional phonon gas model (PGM), which relies on well-defined phonon group velocities and becomes questionable in disordered systems, the GKMA approach uses the Green-Kubo formula combined with lattice dynamics. This allows it to include anharmonic effects to full order without needing to define phonon velocities, making it particularly powerful for studying amorphous materials where periodicity is absent [5] [33]. This application note details the use of GKMA to investigate thermal conductivity in amorphous silicon dioxide (a-SiO₂), a material of widespread technological importance in integrated circuits and microelectronics.
Key Theoretical Concepts and the GKMA Formalism
Vibrational Mode Classification in Amorphous Materials
In amorphous materials, vibrational modes are categorized into three distinct regimes based on their characteristics and heat transport mechanisms:
- Propagons: Low-frequency, wave-like propagating modes similar to those in crystals.
- Diffusons: Mid-frequency, non-propagating modes that carry heat through a diffusive mechanism.
- Localized Modes (Locons): High-frequency modes that are highly spatially localized. Traditionally, these were thought to contribute negligibly to thermal conductivity [5].
The Green-Kubo Modal Analysis (GKMA) Framework
The GKMA method computes thermal conductivity as a direct summation of individual mode contributions. The core equations are as follows:
The total heat flux (J) is obtained from the sum of all individual mode contributions: J = (1/V) * Σₙ [ Eᵢ vᵢ⁽ⁿ⁾ + (1/2) * Σ_{j≠i} (rᵢⱼ · vᵢ⁽ⁿ⁾) Fᵢⱼ ] where n is the mode index, V is the supercell volume, Eᵢ is the energy of atom i, vᵢ⁽ⁿ⁾ is the contribution of mode n to the velocity of atom i, rᵢⱼ is the distance between atoms i and j, and Fᵢⱼ is the force between them [5].
The thermal conductivity tensor (κ) is then given by: κ = (1/(kB T² V)) * Σₙ ∫₀∞ ⟨J⁽ⁿ⁾(t) · J(0)⟩ dt where *kB* is Boltzmann's constant, T is temperature, and the integral represents the heat flux autocorrelation function for mode n [5] [33].
To account for quantum corrections in specific heat, which is crucial for accurate temperature dependence, the following expression is used: κₙ(T) = fQ(T, ωₙ) * fκ(T) * ω(T) where f_Q is the ratio of quantum to classical specific heat, f_κ represents the GKMA-derived mode contributions, and ω is the phonon frequency, which itself can exhibit temperature dependence [5].
Experimental Protocols and Computational Methodologies
GKMA Simulation Protocol for a-SiO₂
The following workflow outlines the primary steps for performing a GKMA calculation, from model preparation to post-processing with quantum corrections.
Structural Model Preparation and Validation
Creating a physically realistic atomic model of a-SiO₂ is crucial for accurate results. The amorphization protocol involves:
- Energy Minimization: Begin with an initial crystalline structure (e.g., α-quartz) and relax it in an NVT ensemble at 5 K for 10 ps [34].
- Melting: Switch to an NPT ensemble and heat the system to a high temperature (e.g., 5000 K) to melt the crystal structure [34].
- Annealing: Slowly cool the molten structure to the target temperature (e.g., 300 K) at a controlled cooling rate (e.g., 1.74 K/ps) to prevent recrystallization [34].
- Validation: Validate the resulting amorphous structure by comparing its Radial Distribution Function (RDF) and Bond Angle Distribution against literature data. Key metrics include the first peak positions of Si-O, O-O, and Si-Si RDFs and the average O-Si-O and Si-O-Si bond angles [34].
Alternative Computational Methods
While GKMA provides deep modal insights, other methods are commonly used to calculate κ:
- Non-Equilibrium Molecular Dynamics (NEMD): A temperature gradient is imposed across the simulation cell, and κ is extracted from Fourier's law: κ = -q / (∂T/∂z), where q is the heat flux. This method is sensitive to system size, especially for thin films [34].
- Approach-to-Equilibrium Molecular Dynamics (AEMD): A periodic thermal profile is created, and the relaxation to equilibrium is monitored. The thermal conductivity is deduced from the relaxation time. This method is computationally efficient and can probe size effects [35].
Key Research Findings and Data Presentation
Modal Contributions to Thermal Conductivity in a-SiO₂
Applying GKMA to a-SiO₂ reveals unexpected contributions from different vibrational modes. The following table quantifies the contributions of propagons, diffusons, and locons across a temperature range.
Table 1: Modal contributions to thermal conductivity in a-SiO₂ using GKMA [5]
| Vibrational Mode Type | Frequency Range | Contribution to κ at 300 K | Contribution to κ at 400-800 K | Primary Transport Mechanism |
|---|---|---|---|---|
| Propagons | Low frequency | ~30% (estimated) | Decreases with temperature | Wave-like propagation |
| Diffusons | Mid-frequency | Dominant contribution | Remains significant | Anharmonic energy diffusion |
| Localized Modes (Locons) | High frequency | <5% (traditional view) | >10% (GKMA result) | Anharmonic interactions & tunneling |
The most significant finding from GKMA is that localized modes contribute more than 10% to the total thermal conductivity in the 400 K to 800 K range and are largely responsible for the increase in κ of a-SiO₂ above room temperature [5]. This overturns the long-held assumption that locons have a negligible contribution due to their spatially confined nature.
Temperature and Size Effects in a-SiO₂ Thin Films
The thermal conductivity of a-SiO₂ exhibits distinct behaviors compared to its crystalline counterpart, especially in thin-film geometries.
Table 2: Temperature and size effects on thermal conductivity of a-SiO₂ [34] [35]
| Material Structure | Temperature Dependence (Typical Range) | Size Effect (Film Thickness) | Representative κ Value at 300 K |
|---|---|---|---|
| Amorphous SiO₂ (Bulk) | Increases above room temperature [5] | Not applicable | ~1.4 W/m·K [34] |
| Amorphous SiO₂ (Thin Film, > 6 nm) | Insensitive from -55 °C to 150 °C [34] | No significant size effect beyond ~4-6 nm [34] [35] | ~1.4 W/m·K [35] |
| Amorphous SiO₂ (Thin Film, < 6 nm) | - | Thermal conductivity decreases with reduced thickness [35] | < 1.2 W/m·K (at 2 nm) [35] |
| Crystalline SiO₂ | Decreases with increasing temperature [34] | Significant size effect; κ ∝ T⁻ᵅ (α = -0.12 to -0.37) [34] | Higher than a-SiO₂ (size-dependent) [34] |
The relationship between system size and thermal conductivity, particularly the critical length scale below which size effects become prominent, can be visualized as follows. This is based on AEMD and NEMD simulation results.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key computational tools and models for studying thermal conductivity in amorphous solids
| Tool / Model / Method | Function / Purpose | Example Application in Field |
|---|---|---|
| Tersoff Potential | Empirical interatomic potential used to describe atomic interactions in SiO₂ and reproduce structural properties of various polymorphs [34]. | NEMD simulations of a-SiO₂ thin films for calculating size-dependent thermal conductivity [34]. |
| Green-Kubo Modal Analysis (GKMA) | A method to directly calculate modal contributions to κ from MD simulations, including full anharmonicity and mode-mode correlations [5] [33]. | Uncovering the non-negligible contribution of localized modes to κ in a-SiO₂ [5]. |
| Non-Equilibrium MD (NEMD) | Imposes a temperature gradient to compute κ directly from Fourier's law. Useful for studying size effects in nanostructures [34]. | Investigating thickness dependence of κ in a-SiO₂ thin films [34]. |
| Approach-to-Equilibrium MD (AEMD) | Calculates κ from the relaxation time of a thermal profile. Computationally efficient for first-principles MD and size effect studies [35]. | Determining the critical length (~6 nm) below which κ decreases in a-SiO₂ [35]. |
| First-Principles Molecular Dynamics (FPMD) | Uses density functional theory (DFT) to compute interatomic forces, offering high accuracy without empirical parametrization [35]. | Generating accurate atomic models of a-SiO₂ and calculating κ with high predictive power [35]. |
| Inverse Participation Ratio (IPR) | A metric used to quantify the degree of spatial localization of a vibrational mode [5]. | Classifying modes as propagons, diffusons, or locons in the analysis of a-SiO₂ [5]. |
The application of Green-Kubo Modal Analysis to amorphous silicon dioxide has fundamentally altered the understanding of thermal transport in disordered materials. The key insight that localized modes contribute significantly to thermal conductivity, especially at elevated temperatures, opens new avenues for materials design. This understanding, framed within the broader context of GKMA for disordered solids, provides researchers with a powerful protocol to dissect and influence thermal transport. This is particularly relevant for the design of thermal barrier coatings, thermoelectric materials with optimized diffuson transport [36], and advanced dielectric layers in microelectronics where precise thermal management is critical for device performance and reliability. The methodologies and findings outlined here serve as a robust template for extending this analysis to other amorphous and nanostructured materials.
Optimizing GKMA Simulations: Addressing Uncertainty and Convergence
Green-Kubo Modal Analysis (GKMA) is an advanced computational formalism that enables the direct calculation of individual phonon or normal mode contributions to the thermal conductivity of materials. A significant advantage of GKMA is its generality; it can be applied to a wide range of solid types, including amorphous materials, crystalline solids, crystalline alloys, and polymers, within a single unified framework [3]. The method operates within the broader context of the Green–Kubo relations, which, in molecular dynamics (MD) simulations, relate the macroscopic thermal conductivity (λ) to the microscopic time correlation of the heat flux vector J(t) in a system at equilibrium [12]. The foundational equation is:
λ = (V / (3kBT^2)) ∫0∞ ⟨J(t) · J(0)⟩ dt
where V is the system volume, T is the temperature, and kB is the Boltzmann constant [12]. The core quantity in this relation, the heat flux auto-correlation function (HFACF), is inherently a statistical measure. In practice, the HFACF is computed as an average over a finite simulation time and a finite number of initial time origins, making the resulting thermal conductivity value subject to statistical uncertainty. For disordered solids such as amorphous materials, this challenge is particularly acute. The complex, non-periodic atomic structures in these materials lead to heat flux correlations that decay more slowly and exhibit more noise compared to crystalline systems. Consequently, achieving adequate sampling of the phase space is computationally expensive, and the statistical uncertainty in the computed thermal conductivity can be significant, potentially obscuring meaningful physical interpretation and limiting the reliability of the results for materials design.
Quantifying and Analyzing Uncertainty
The statistical uncertainty in GKMA simulations manifests primarily in the calculation of the HFACF and its time integral. Key metrics for quantifying this uncertainty are summarized in Table 1.
Table 1: Key Quantitative Metrics for Statistical Uncertainty in GKMA Simulations
| Metric | Typical Range/Value | Interpretation and Impact |
|---|---|---|
| HFACF Decay Time | Varies by material (e.g., longer in disordered solids) | The time required for the HFACF to decay to zero. Longer decay requires longer simulation time to capture the full integral [12]. |
| Standard Error of λ | Often reported as a percentage of λ | A measure of the convergence of the cumulative integral. A common criterion is a variation of less than 5-10% over a significant portion of the plateau region. |
| Correlation Length | System-size dependent | The size of the simulation cell must be larger than the relevant correlation lengths for heat transport to avoid finite-size effects. |
| Sampling Frequency | Sufficient to capture atomic vibrations | The time interval for saving the heat flux data must be fine enough to resolve the fastest atomic motions contributing to J(t). |
The following diagram illustrates the logical workflow for quantifying statistical uncertainty in a GKMA simulation, from running the molecular dynamics simulation to analyzing the convergence of the thermal conductivity.
Protocol for Uncertainty Quantification
Objective: To determine the thermal conductivity (λ) of a disordered solid using GKMA and reliably estimate its statistical uncertainty.
Materials and Computational Setup:
- Software: A molecular dynamics package with Green-Kubo capability, such as LAMMPS [12].
- Force Field: A suitable interatomic potential for the material under study (e.g., OPLS-AA for organic molecules, other empirical potentials or machine-learning potentials for inorganic glasses) [12].
- Initial Structure: A realistically modeled atomic structure of the disordered solid, typically generated through a melt-and-quench procedure in MD.
- Computational Resources: High-performance computing (HPC) resources, as long simulation times are required for adequate sampling.
Procedure:
- System Equilibration:
- Energy minimize the initial atomic structure.
- Equilibrate the system in the NPT ensemble (constant Number of particles, Pressure, and Temperature) at the target temperature and pressure for a sufficient duration to stabilize density.
- Further equilibrate in the NVE ensemble (constant Number of particles, Volume, and Energy) to ensure stable energy fluctuations.
Production Run and Data Sampling:
- Run a long NVE simulation while saving the full heat flux vector J(t) at regular intervals. The sampling frequency, Δt, must be short enough to capture atomic vibrations (e.g., 1-5 femtoseconds).
- The total simulation time (ttotal) should be several times longer than the characteristic decay time of the HFACF. For disordered solids, this may require simulations spanning nanoseconds.
Heat Flux Correlation Analysis:
- From the saved J(t) data, compute the ensemble-averaged HFACF, C(t) = ⟨J(t) · J(0)⟩.
- The ensemble average is performed by splitting the time series into multiple, possibly overlapping, segments (block averaging).
Thermal Conductivity Integration and Uncertainty Estimation:
- Compute the time-dependent thermal conductivity as λ(τ) = (V / (3kBT^2)) ∫0τ C(t) dt.
- Plot λ(τ) as a function of the upper integration limit τ. A well-converged result will show a clear plateau.
- Uncertainty Estimation: The standard error of λ can be estimated by dividing the production run into several independent blocks, calculating λ for each block, and then computing the standard deviation of the mean of these block values. The result is considered converged when this standard error is below a pre-defined threshold (e.g., <10% of the mean λ).
Sampling Strategies for Disordered Solids
Achieving sufficient sampling is the primary challenge in obtaining reliable GKMA results for disordered solids. The following protocol outlines a strategic approach to enhance sampling efficiency.
Protocol for Enhanced Sampling
Objective: To implement advanced sampling strategies that reduce the statistical uncertainty in the HFACF without prohibitively increasing computational cost.
Procedure:
- Extended Simulation Time:
- The most straightforward method. For a fixed system size, the statistical error in λ decreases approximately as 1/√(ttotal). For disordered solids, multi-nanosecond simulations are often the baseline requirement.
Multiple Independent Trajectories:
- Instead of one very long simulation, run several independent simulations from different initial conditions (e.g., different velocity seeds or slightly different equilibrated structures).
- Calculate λ from each trajectory and report the final result as the mean and standard deviation across all runs. This provides a more robust estimate of statistical uncertainty.
Heat Flux Component Sampling (GKMA Breakdown):
- Leverage the GKMA formalism to break down the total heat flux into contributions from different atom types or modes [3] [12].
- As shown in the diagram and table below, this involves decomposing the virial component of the heat flux, Jv, into contributions from specific atomic pairwise interactions (e.g., C-C, O-O, O-H) [12].
- This decomposition not only provides physical insight but also allows for targeted analysis. One can monitor the convergence of specific cross-correlation terms, which may require different sampling strategies than the total correlation.
Table 2: Research Reagent Solutions for GKMA Sampling
| Item / "Reagent" | Function in Protocol |
|---|---|
| Long-Timescale MD Software (LAMMPS) | Core computational engine for performing the equilibrium molecular dynamics simulations and calculating the heat flux [12]. |
| Validated Force Field | Defines the interatomic interactions, which are critical for accurately modeling the potential energy and forces that govern the heat flux [12]. |
| Structure Generator (e.g., packmol, in-house scripts) | Creates realistic initial atomic configurations of the disordered solid for simulation. |
| Heat Flux Decomposition Script | Custom code (e.g., for LAMMPS) that breaks down the total heat flux into modal or atomic-pair contributions as per the GKMA formalism [3] [12]. |
| Correlation Function Analyzer | Scripts (e.g., in Python/MATLAB) to compute the HFACF, its integral, and perform block-averaging for error analysis. |
The following diagram illustrates the logical relationship during the decomposition of the heat flux for more detailed sampling, as enabled by GKMA and related methods.
An Applied Case Study Analysis
The application of atomic-level breakdown, conceptually similar to the principles of GKMA, is illustrated in a study on liquid alcohols [12]. This study provides a valuable template for analyzing disordered solids. Researchers decomposed the virial heat flux Jv into components from specific atomic pairwise interactions (e.g., Carbon-Carbon, Oxygen-Hydrogen) [12]. The thermal conductivity was correspondingly broken down into auto-correlation and cross-correlation terms between these atomic pairs:
λvv = ΣX,Y λX-Y = ΣX,Y (V / (3kBT^2)) ∫0∞ ⟨JX(t) · JY(0)⟩ dt
where X and Y represent atom types [12]. The analysis revealed non-trivial cross-correlations; for instance, in ethylene glycol, a negative cross-correlation between O-H and C-O interactions (λOH-CO) indicated a competitive relationship in their contribution to overall thermal transport [12]. From a sampling perspective, this finding is critical. It implies that the convergence of the total λ is not merely the sum of independently converging components. The sampling time must be long enough to capture the accurate statistical relationship between these cross terms, which fluctuate and decay at different rates. Failure to do so can lead to an incorrect estimation of both the magnitude and the physical mechanism of heat conduction. This case study underscores that sophisticated sampling is not just a numerical exercise but is essential for uncovering the true nano-scale physics of thermal transport in disordered systems.
Quantifying Uncertainty in Current Autocorrelation Functions (CAFs)
In the context of Green-Kubo Modal Analysis (GKMA) for thermal conductivity research in disordered solids, the Current Autocorrelation Function (CAF) serves as a fundamental quantity. The Green-Kubo relation establishes that thermal conductivity (κ) is proportional to the time integral of the heat current autocorrelation function [3]. For disordered materials, where traditional phonon gas models are insufficient, accurately quantifying the uncertainty in these autocorrelation functions becomes paramount for reliable thermal conductivity predictions. The CAF measures how heat currents in a material remain correlated over time, and its decay profile reveals critical information about thermal transport mechanisms.
Uncertainty quantification in CAFs is essential because these functions are computed from molecular dynamics simulations with inherent stochastic variations. The precision of your GKMA results depends directly on understanding and minimizing these uncertainties, particularly for disordered solids where heterogeneous atomic structures create complex, non-exponential decay patterns that challenge conventional analysis methods.
Theoretical Foundation of ACF Uncertainty
Autocorrelation Function Formalism
For a discrete time series of heat current observations J(t) with N measurements, the sample Current Autocorrelation Function is calculated as [37] [38]:
[ \hat{\rho}(k) = \frac{\sum{t=1}^{N-k} (J(t) - \bar{J})(J(t+k) - \bar{J})}{\sum{t=1}^{N} (J(t) - \bar{J})^2} ]
where k is the lag time, (\bar{J}) is the mean heat current, and (\hat{\rho}(k)) represents the estimated correlation at lag k. In GKMA applications, this function is computed for individual vibrational modes to determine their specific contributions to thermal conductivity.
The theoretical ACF for a stationary process is defined as (\rho(k) = \text{corr}(J(t), J(t+k))), but the sample ACF (\hat{\rho}(k)) deviates from this true value due to finite sampling. For white noise processes, the sample ACF values are asymptotically independent and normally distributed under ideal conditions, but recent research has revealed significant deviations from normality beyond specific lags, challenging traditional assumptions [39].
- Finite Sampling Error: Molecular dynamics simulations provide limited time series data, making CAF estimates susceptible to statistical fluctuations.
- Measurement Imperfections: In computational experiments, force field inaccuracies and numerical integration errors affect current correlations [37].
- Non-Normality of ACF Distributions: Empirical studies show that ACF values at larger lags deviate from normal distribution assumptions, particularly for the residuals of time series models [39].
- Correlation Between Successive ACFs: The sum of all sample ACFs must equal -½ according to Hassani's theorem, creating dependencies between ACF values at different lags that violate independence assumptions [39].
Methodologies for Uncertainty Quantification
Monte Carlo Uncertainty Propagation
Monte Carlo methods provide a robust approach for quantifying uncertainty in CAFs by accounting for both measurement imperfections and finite sampling effects. The procedure involves [37]:
- Resampling: For each original heat current time series, generate a large number (typically 1000-10000) of synthetic series where each data point has a probability (1-F) of being "flipped" or perturbed according to estimated measurement reliability.
- CAF Calculation: Compute the autocorrelation function for each resampled series.
- Distribution Analysis: For each lag k, build a distribution of correlation values from the resampled CAFs.
- Confidence Interval Estimation: Determine confidence intervals (typically 95%) for each lag's correlation value from the resampled distributions.
This approach directly propagates uncertainty from the original measurements through the entire CAF calculation process, providing empirical confidence intervals that do not rely on potentially invalid normality assumptions.
Standard Error Estimation from Multiple Realizations
When you have performed multiple independent simulations (e.g., 25 experiments as mentioned in the search results), you can estimate standard errors through [37]:
- Individual CAF Calculation: Compute separate CAFs for each independent simulation.
- Mean CAF Determination: Calculate the mean CAF across all realizations.
- Standard Error Calculation: For each lag k, compute the standard error as (\text{SE}(k) = \sigma(k)/\sqrt{N}), where (\sigma(k)) is the standard deviation of CAF values at lag k across the N realizations.
This approach is particularly valid when CAF values at different lags are approximately independent, though this assumption may break down for larger lags where correlations between successive ACF values become significant [39].
Analytical Approaches
For white noise processes with finite fourth moments, the theoretical distribution of sample ACF values is asymptotically normal with variance approximately 1/N under the null hypothesis of no correlation [39]. However, this analytical approach has limitations for CAFs in disordered solids, where significant correlations exist at short time scales. The analytical confidence bands for a true autocorrelation of zero are given by (\pm z{\alpha/2}/\sqrt{n}), where (z{\alpha/2}) is the critical value from the normal distribution (typically 1.96 for 95% confidence) [38].
Table 1: Comparison of Uncertainty Quantification Methods for CAFs
| Method | Requirements | Advantages | Limitations |
|---|---|---|---|
| Monte Carlo Propagation | Estimate of measurement reliability (F₀, F₁) | Accounts for measurement imperfections; Makes minimal distribution assumptions | Computationally intensive; Requires reliability estimates |
| Standard Error from Realizations | Multiple independent simulations | Simple to implement; Uses actual observed variance | Requires substantial computational resources for multiple runs |
| Analytical Confidence Intervals | Large sample size; Stationary process | Computationally efficient; Well-established theory | Relies on potentially invalid normality assumptions; Less accurate for small samples |
Experimental Protocol for CAF Uncertainty Analysis
Prerequisite Data Collection
- Perform Molecular Dynamics Simulations: Run sufficiently long NVE or NVT simulations of your disordered solid system to generate heat current trajectories.
- Extract Heat Current Time Series: Calculate the heat current vector J(t) for each timestep throughout the simulation duration.
- Ensure Stationarity: Verify that the statistical properties of the heat current time series do not exhibit drift or non-stationarity using unit root tests or visual inspection of the ACF decay [40].
Step-by-Step Uncertainty Quantification Protocol
Compute Raw CAF:
- For each independent simulation, calculate the current autocorrelation function using the standard formula.
- Normalize by the zero-time correlation to obtain a normalized CAF decaying from 1 to 0.
Apply Monte Carlo Uncertainty Propagation:
- For each original J(t) series, generate 5000 synthetic series by applying random perturbations according to your estimated measurement reliability.
- Compute CAFs for all synthetic series.
- For each lag k, determine the 2.5th and 97.5th percentiles of the correlation values to establish 95% confidence intervals.
Calculate Standard Errors Across Realizations:
- If multiple independent simulations are available, compute the mean and standard deviation of CAF values at each lag across realizations.
- Determine standard errors as (\text{SE}(k) = \sigma(k)/\sqrt{M}) where M is the number of realizations.
Validate with Hassani's Theorem:
- Compute the sum of all sample ACF values across lags to verify it approximates -½ as required by theory [39].
- Significant deviations may indicate issues with stationarity or insufficient sampling.
Assess Normality of ACF Distributions:
- For the CAF values at each lag across multiple realizations, perform normality tests (e.g., Shapiro-Wilk test) to verify distributional assumptions.
- Pay particular attention to larger lags where deviations from normality are more pronounced [39].
Integrate Uncertainty in GKMA Analysis:
- Propagate the CAF confidence intervals through the Green-Kubo integration to determine uncertainty in thermal conductivity predictions.
- Use weighted fitting procedures that account for CAF uncertainties when fitting decay models.
Diagram 1: Experimental workflow for quantifying uncertainty in Current Autocorrelation Functions within GKMA framework.
Research Reagent Solutions
Table 2: Essential Computational Tools for CAF Uncertainty Analysis
| Tool/Software | Function | Application in CAF Uncertainty |
|---|---|---|
| LAMMPS | Molecular Dynamics Simulator | Generates heat current trajectories from atomic simulations of disordered solids |
| NumPy/SciPy | Numerical Computing Libraries | Implements CAF calculation and statistical analysis procedures |
| Custom Monte Carlo Code | Uncertainty Propagation | Performs resampling and confidence interval estimation for CAFs |
| Statsmodels (Python) | Statistical Testing | Provides normality tests and additional statistical validation |
| MATLAB/GNU Octave | Alternative Numerical Analysis | Optional platform for time series analysis and ACF computation |
Data Presentation and Interpretation
Structured Data Representation
When presenting CAF uncertainty results, use clearly structured tables that include:
Table 3: Example CAF Uncertainty Quantification Results for Amorphous Silicon at 300K
| Lag (ps) | Mean CAF | Monte Carlo 95% CI Lower | Monte Carlo 95% CI Upper | Standard Error | Normality p-value |
|---|---|---|---|---|---|
| 0.1 | 0.892 | 0.885 | 0.899 | 0.0037 | 0.243 |
| 0.5 | 0.641 | 0.627 | 0.655 | 0.0072 | 0.118 |
| 1.0 | 0.352 | 0.331 | 0.373 | 0.0108 | 0.065 |
| 2.0 | 0.128 | 0.104 | 0.152 | 0.0123 | 0.017 |
| 5.0 | 0.021 | -0.008 | 0.045 | 0.0137 | 0.003 |
Interpretation Guidelines
- Significant Correlations: CAF values whose confidence intervals do not include zero indicate statistically significant current correlations at that time lag.
- Uncertainty Growth: Typically, uncertainty in CAF estimates increases with lag time due to fewer data points contributing to the calculation.
- Non-Normality Indicators: Low p-values in normality tests (typically <0.05) at larger lags confirm theoretical expectations of distributional deviations [39].
- Integration Impact: Lags with larger relative uncertainty contribute more to the overall uncertainty in thermal conductivity values obtained through Green-Kubo integration.
Validation and Troubleshooting
Quality Control Measures
- Hassani's Theorem Validation: Regularly check that the sum of all sample ACF values approximates -½; significant deviations may indicate issues with data stationarity or calculation errors [39].
- Consistency Across Methods: Compare uncertainty estimates from Monte Carlo methods with those from multiple realizations; large discrepancies may signal methodological issues.
- Convergence Testing: Verify that CAF estimates and their uncertainties stabilize with increasing simulation duration and number of Monte Carlo samples.
Common Issues and Solutions
Problem: Excessively wide confidence intervals at all lags.
- Solution: Increase simulation duration to improve statistical sampling of heat current fluctuations.
Problem: Significant deviations from Hassani's theorem.
- Solution: Check for non-stationarity in heat current time series and apply appropriate detrending methods.
Problem: Strong non-normality in ACF distributions even at small lags.
- Solution: Consider non-parametric confidence intervals based on percentiles rather than normal distribution assumptions.
Problem: Discrepancies between uncertainty estimates from different methods.
- Solution: Prioritize Monte Carlo approaches when measurement reliability estimates are available, as they more comprehensively account for error sources.
Strategies for Achieving Convergence in Long-Tail Integrals
Within the framework of Green-Kubo Modal Analysis (GKMA) for thermal conductivity research in disordered solids, the challenge of achieving convergence in long-tail integrals represents a significant computational hurdle. This article details application notes and protocols for overcoming this challenge, focusing on the precise calculation of the heat current autocorrelation function (HCACF) integral. We provide a structured overview of convergence strategies, quantitative data presentation on key parameters, and step-by-step experimental protocols validated through application to materials like amorphous silicon dioxide (a-SiO2). By implementing these strategies, researchers can obtain reliable, reproducible thermal conductivity data for disordered systems, enabling advancements in thermal management and material design for applications ranging from semiconductor devices to thermal energy storage.
The Green-Kubo Modal Analysis (GKMA) method computes thermal conductivity (κ) by projecting the total heat flux onto individual vibrational modes and summing their contributions to the Green-Kubo integral [41] [42]. For a system with N atoms, the total thermal conductivity is given by: κ = Σn=13N κ(n)
where the contribution of mode n is: κ(n) = (1/kBT2V) ∫0∞ 〈Jn(0)Jtotal(t)〉 dt
Here, Jn is the heat flux contribution from mode n, and the integral represents the long-tail correlation between modal and total heat flux [41]. In disordered solids such as amorphous silicon dioxide (a-SiO2), the HCACF often exhibits a slow decay, leading to the "long-tail integral" problem. Failure to properly converge this integral results in inaccurate thermal conductivity predictions and an incomplete physical picture of thermal transport mechanisms [41]. For instance, in a-SiO2, converged GKMA simulations revealed that localized modes (locons) contribute more than 10% to the total thermal conductivity above room temperature—an effect previously overlooked due to inadequate convergence [41].
Quantitative Data for Convergence Control
Achieving convergence requires careful control of multiple simulation parameters. The following tables summarize the key quantitative factors and their impact on the convergence of the long-tail integral.
Table 1: Critical Simulation Parameters for GKMA Convergence
| Parameter | Description | Typical Value/Range | Effect on Convergence |
|---|---|---|---|
| Supercell Size | Number of atoms (N) in the simulation cell [41]. | 384 - 3840 atoms [41] [31] | Larger sizes minimize finite-size effects and allow more low-frequency modes to be captured. |
| Correlation Time | Upper limit (tmax) for the HCACF integral [41]. | 10 - 100 ps [41] | Must be long enough for the HCACF to fully decay to zero. Insufficient tmax truncates the integral. |
| Sampling Statistics | Number of independent simulation runs and correlation samples per run. | 5-10 independent runs [31] | Averages out statistical noise in the HCACF, smoothing the integral. |
| Quantum Correction | Factor fQ applied to mode TC to correct for classical MD occupations [41]. | fQ = CQ/CCL | Essential for accurate temperature dependence, especially below the Debye temperature. |
Table 2: Convergence Indicators and Diagnostics
| Diagnostic | Target Behavior | Implication for Convergence |
|---|---|---|
| HCACF Decay | HCACF should decay to zero within the simulation correlation time [41]. | A non-decaying tail indicates insufficient correlation time or system size. |
| Cumulative Integral Plateau | The value of the running integral κ(t) = ∫0t 〈J(0)J(t')〉 dt' must reach a stable plateau [41]. | Fluctuations or a persistent slope at long times signal lack of convergence. |
| Modal Contribution Summation | The sum of individual mode contributions Σκ(n) should converge to the total κ from direct Green-Kubo [42]. | Large discrepancies indicate poor sampling or an incomplete modal basis. |
Experimental Protocols for GKMA
This section provides a detailed, step-by-step protocol for performing a converged GKMA simulation, using the example of a hexagonal boron nitride (hBN) sheet as referenced in the search results [31].
The following diagram illustrates the complete GKMA workflow, from structure preparation to the analysis of converged thermal conductivity.
Step-by-Step Procedure
Step 1: Structure Creation and Energy Minimization
- Protocol: Begin by creating an atomic structure file (e.g.,
xyz.in). For a 2D material like hBN, a structure of ~10x10 nm² (e.g., 3840 atoms) is a reasonable starting point [31]. The potential file (e.g.,BN_Sevik_2011.txtfor hBN) must be specified. - Convergence Tip: Energy minimization is crucial to ensure atoms are at stable equilibrium positions before the phonon calculation. Use a steepest descent algorithm with a force tolerance of 1e-8 eV/Å and a maximum of 10,000 iterations [31].
- Verification: Check the output to confirm that the force tolerance was met.
Step 2: Phonon Calculation and Eigenvector Extraction
- Protocol: Perform a gamma-point phonon calculation on the entire supercell using the
compute_phononkeyword. A cutoff of 4.0 Å and a displacement of 0.005 Å are typical parameters [31]. This calculation outputs theeigenvector.outfile. - Convergence Tip: The phonon cutoff must be large enough to include all relevant atomic interactions (e.g., first and second nearest neighbors). Rename
eigenvector.outtoeigenvector.infor use in subsequent GKMA simulations. - Verification: Use tools like
gpyumd.load.load_frequency_infoto plot the vibrational density of states and ensure the frequency spectrum is physically reasonable [31].
Step 3: Molecular Dynamics Equilibrium
- Protocol: Equilibrate the minimized structure in an NVT (canonical) ensemble at the target temperature (e.g., 300 K) using a Nosé-Hoover thermostat for a duration sufficient to stabilize temperature and energy (typically 0.5-1 million steps with a 1 fs timestep).
- Convergence Tip: The equilibration phase should be long enough to erase any memory of the initial configuration.
Step 4: HNEMA/GKMA Production Run
- Protocol: Run an NVE (microcanonical) simulation using the
hnemaorgkmakeyword, which reads theeigenvector.infile. This projects the atomic velocities onto the normal modes and records the modal contributions to the heat current. A production run of 10-100 million steps is typical [31]. - Convergence Tip: To improve statistics, run multiple (e.g., 5-10) independent simulations with different initial random velocity seeds. The
hnemamethod is often computationally more efficient thangkmafor initial tests [31].
Step 5: HCACF Calculation and Integral Evaluation
- Protocol: Calculate the total and modal HCACF from the heat flux data. Compute the running integral κ(t) = (1/kBT2V) ∫0t 〈J(0)J(t')〉 dt' for the total and cumulative modal contributions.
- Convergence Tip: The integral must be computed to a time tmax where the HCACF has fully decayed to zero. The running integral κ(t) should exhibit a clear plateau. If it does not, increase the correlation time or the total simulation length.
Step 6: Quantum Correction and Final Analysis
- Protocol: Apply a quantum correction to the mode-specific thermal conductivity. The correction factor for mode n with frequency ω is fQ = CQ(ω, T)/CCL, where CQ and CCL are the quantum and classical specific heats, respectively [41]. The final, quantum-corrected thermal conductivity is κcorrected(n) = fQ κMD(n).
- Convergence Tip: This step is vital for obtaining agreement with experimental data, especially at temperatures below the material's Debye temperature [41].
The Scientist's Toolkit
The following table lists essential "research reagents" — computational tools and inputs — required to perform a GKMA simulation successfully.
Table 3: Key Research Reagent Solutions for GKMA
| Item Name | Function/Description | Critical Specifications |
|---|---|---|
| Interatomic Potential | Defines the forces between atoms. | Must be appropriate for the material and its phase (e.g., Tersoff for hBN [31], ReaxFF for polymers [41]). |
Eigenvector File (eigenvector.in) |
Contains the normal mode eigenvectors that form the basis for modal decomposition. | Generated from a gamma-point phonon calculation on the entire, minimized supercell [31]. |
| Equilibrated Configuration | The starting atomic positions and velocities for the production NVE run. | Obtained from a prior NVT simulation, with stable temperature and potential energy. |
| Quantum Correction Script | Applies the necessary temperature-dependent specific heat correction to MD results. | Must calculate fQ = CQ/CCL for each mode frequency [41]. |
Troubleshooting and Validation
Common Convergence Issues and Solutions:
- No Plateau in Cumulative Integral: This is the most direct sign of a non-converged long-tail integral. The solution is to increase the correlation time (tmax) and/or increase the sampling statistics by running more independent simulations [41].
- High-Frequency Noise in HCACF: This is often a result of insufficient sampling. Increase the number of independent runs and ensure the production phase is long enough to capture the slowest decaying correlations.
- Unphysical Modal Contributions: If the contributions from individual modes are unphysical (e.g., strongly negative), verify the correctness of the eigenvector calculation and ensure the energy minimization reached a true local minimum.
Validation Against Experimental Data: The ultimate validation of a converged GKMA simulation is comparison with experimental thermal conductivity data. For example, the protocol above, when applied to amorphous silicon (a-Si), has shown "the best agreement with experiments to date" [42]. Similarly, for a-SiO2, a converged GKMA simulation captured the experimentally observed increase in thermal conductivity above room temperature, which was linked to the significant contribution from localized modes [41]. This level of agreement confirms that the strategies for achieving convergence in the long-tail integrals are effective and reliable.
The Role of Simulation Length and System Size
Accurate prediction of thermal conductivity (TC) in disordered solids, such as amorphous polymers and inorganic glasses, is critical for applications ranging from thermal insulation to electronics packaging. Green-Kubo Modal Analysis (GKMA) has emerged as a powerful method for resolving the contributions of individual vibrational modes to heat transfer in these complex materials [43] [15]. Unlike traditional approaches based on the phonon gas model, GKMA provides a unified framework that captures the contributions of propagating, diffusive, and localized modes, all within a formalism that fully incorporates anharmonic effects [43].
The reliability of GKMA predictions, however, depends critically on two computational parameters: simulation length and system size. Statistical convergence of the heat current autocorrelation function requires sufficiently long molecular dynamics (MD) trajectories, while the system size must be large enough to capture the relevant physics of vibrational modes, particularly localized modes (locons) that dominate thermal transport in many disordered solids [43]. This Application Note provides detailed protocols for determining appropriate simulation dimensions to ensure accurate thermal conductivity predictions using GKMA.
Theoretical Foundation of GKMA
The GKMA method decomposes the total thermal conductivity into contributions from individual vibrational modes, providing unprecedented insight into heat carriers in disordered solids [15]. The fundamental GKMA equation for the running thermal conductivity of mode n is given by:
[\kappa{xx,n}(t) = \frac{1}{kBT^2V} \int0^{t} dt' \langle J{x,n}(t')J_{x}(0)\rangle]
where (k{\rm B}) is Boltzmann's constant, (T) is temperature, (V) is system volume, (J{x}(0)) is the total heat current at time zero, and (J_{x,n}(t')) is the mode-specific heat current at time (t') [11]. The angle brackets denote averaging over different time origins.
This formulation allows researchers to quantify contributions from different classes of vibrational modes—propagons, diffusons, and locons—each exhibiting distinct spatial characteristics and transport mechanisms [43]. The participation ratio (PR) is commonly used to distinguish these modes, with locons typically defined as having PR < 0.1-0.2 [43].
System Size Considerations
Minimum Size Requirements
System size must be sufficient to accommodate the longest wavelength vibrations and properly represent material structure. For disordered solids, this often means capturing the inherent structural heterogeneity.
Table 1: System Size Recommendations for Different Material Classes
| Material Class | Minimum Atoms | Key Considerations | Representative Studies |
|---|---|---|---|
| Amorphous Polymers (PMMA, PS, PVC) | 1000-2000 atoms | Must capture inter-chain and intra-chain interactions; locons dominate (90%+ of modes) [43] | 89.7-94.9% of modes classified as locons (PR<0.1) [43] |
| Amorphous Inorganics (a-SiO₂) | 1000+ atoms | Must represent medium-range disorder; locons contribute >10% to TC [15] | Significant locon contributions (10%+) from 400-800K [15] |
| Lennard-Jones Argon | 1000 atoms | Smaller systems sufficient for simple potentials [44] | GK method gives size-independent results with <1000 atoms [44] |
| Stillinger-Weber Silicon | 1000 atoms | Comparable to LJ argon for GK method [44] | Size-independent results with <1000 atoms [44] |
Size Effect Phenomena
Insufficient system size can lead to significant underestimation of thermal conductivity, particularly when using non-equilibrium methods. For Stillinger-Weber silicon at 500 K, the linear extrapolation procedure applied to direct method results can underpredict the GK thermal conductivity by a factor of 2.5 when minimum system sizes are too small [44]. The GK method itself generally demonstrates faster convergence with system size compared to non-equilibrium approaches [44].
Simulation Length Requirements
Statistical Convergence
Long simulation times are essential for accurate calculation of heat current autocorrelation functions, particularly for systems with slow dynamics such as ionic liquids and polymers.
Table 2: Simulation Length Guidelines for Different System Types
| System Type | Minimum Production Time | Correlation Time | Statistical Sampling | Special Considerations |
|---|---|---|---|---|
| Protic Ionic Liquids | 50+ ns | Extensive correlation times required due to sluggish dynamics [2] | Multiple independent trajectories recommended [2] | Coulombic interactions slow dynamics; polarization effects important [2] |
| Amorphous Polymers | 10+ ns | Dependent on modal lifetimes | Time-origin averaging essential | Anharmonic interactions between chains crucial [43] |
| Amorphous Silicon | Simulation-dependent | Case-dependent correlation times | Standard time-origin averaging | Quantum corrections needed for low-temperature predictions [15] |
Uncertainty Quantification
Modern approaches such as the KUTE algorithm address the challenge of statistical uncertainty in Green-Kubo calculations [2]. The running thermal conductivity can be calculated as a weighted average:
[\gammai^{\alpha\beta} = \frac{\sum{k=i}^N Ik^{\alpha\beta}/u^2(Ik^{\alpha\beta})}{\sum{k=i}^N u^{-2}(Ik^{\alpha\beta})}]
where (Ik^{\alpha\beta}) is the running integral of the correlation function at time step *k*, and (u(Ik^{\alpha\beta})) is its statistical uncertainty [2]. This approach eliminates arbitrary cutoffs by identifying plateaus in the running transport coefficient where all points are statistically equivalent within uncertainty [2].
Comprehensive GKMA Protocol
The following diagram illustrates the complete GKMA methodology, integrating both lattice dynamics and molecular dynamics components:
Step-by-Step Implementation
System Preparation
- Begin with a well-relaxed atomic structure that accurately represents the disordered material.
- Perform energy minimization with tolerance of 10 kJ/mol or equivalent [2].
- Conduct NpT ensemble equilibration for 10+ ns to stabilize density [2].
- Follow with NVT ensemble equilibration for 5+ ns at target temperature [2].
Lattice Dynamics Calculation
Molecular Dynamics Simulation
Modal Analysis
- Project atomic velocities onto normal mode eigenvectors: (\boldsymbol{v}i (t) = \frac{1}{\sqrt{mi}} \sum{n} \boldsymbol{e}{i,n} \cdot \boldsymbol{\dot{X}}_n(t)) [11].
- Compute modal heat currents: (\boldsymbol{J}^{\text{pot}}n = \left(\sum{i} \frac{1}{\sqrt{mi}} \mathbf{W}i \cdot \boldsymbol{e}{i,n} \right) \cdot \boldsymbol{\dot{X}}n(t)) [11].
- Calculate heat current autocorrelation function with time-origin averaging.
Thermal Conductivity Calculation
- Integrate autocorrelation function using Green-Kubo formula.
- Apply quantum correction for specific heat when comparing to low-temperature experiments: (\kappa{\text{corrected}} = fQ \cdot f\kappa(T) \cdot \omega(T)) where (fQ) is the quantum-to-classical specific heat ratio [15].
- Use uncertainty quantification methods (e.g., KUTE algorithm) to identify convergence plateau [2].
The Scientist's Toolkit
Table 3: Essential Computational Tools for GKMA Implementation
| Tool Name | Function | Application Notes |
|---|---|---|
| GPUMD | Molecular dynamics simulator with built-in GKMA support | Specialized for thermal transport; computes potential part of heat current decomposition [11] |
| KUTE | Python package for Green-Kubo uncertainty quantification | Implements weighted averaging; eliminates arbitrary cutoffs in correlation integrals [2] |
| OpenMM | MD simulation toolkit | Supports polarizable force fields; used for IL studies [2] |
| LAMMPS | General-purpose MD simulator | Compatible with various analysis packages for post-processing |
| CL&Pol Force Field | Polarizable force field for ionic liquids | Captures hydrogen bonding and polarization effects [2] |
| ReaxFF | Reactive force field | Suitable for studying polymer chains with multi-body potentials [15] |
Critical Considerations for Disordered Solids
Unique Characteristics of Amorphous Polymers
Amorphous polymers exhibit exceptionally high populations of localized modes (89.7-94.9% with PR < 0.1) compared to amorphous inorganic materials [43]. These locons contribute significantly to overall thermal conductivity (over 80% in studied thermoplastics), contrary to traditional assumptions that localized modes have negligible contribution [43]. System size must be sufficient to capture inter-chain interactions, as heat conduction predominantly occurs when localized modes involve two chains [43].
Temperature Dependence Considerations
For amorphous silica, GKMA reveals that locons contribute more than 10% to thermal conductivity between 400-800 K and are largely responsible for the increase in thermal conductivity above room temperature [15]. This temperature-dependent behavior necessitates simulations at multiple temperatures with appropriate quantum corrections:
[\kappa{\text{corrected}} = fQ \cdot f_\kappa(T) \cdot \omega(T)]
where (fQ) represents the quantum-to-classical specific heat ratio, (f\kappa(T)) the GKMA-derived mode contributions, and (\omega(T)) the temperature-dependent phonon frequencies [15].
Proper selection of simulation length and system size is paramount for accurate thermal conductivity prediction in disordered solids using GKMA. System sizes of 1000+ atoms are typically sufficient for Green-Kubo methods, while simulation lengths must be extended for systems with slow dynamics such as ionic liquids and polymers. The unique dominance of localized modes in amorphous polymers necessitates particular attention to system size to properly capture inter-chain interactions. Implementation of uncertainty quantification methods such as KUTE provides robust criteria for correlation function convergence, eliminating arbitrary cutoffs and improving the reliability of GKMA predictions for disordered materials.
Leveraging Advanced Algorithms for Robust GKMA Calculations
Green-Kubo Modal Analysis (GKMA) is an advanced computational formalism that enables the direct calculation of individual phonon or normal mode contributions to the thermal conductivity of materials. This method represents a significant advancement in the field of thermal transport physics as it provides a unified framework applicable to a vast range of materials, including crystalline solids, amorphous materials, random alloys, polymers, and even molecules [3] [8]. The foundational principle of GKMA lies in its combination of lattice dynamics formalism with the Green-Kubo formula for thermal conductivity, resulting in a formulation where thermal conductivity becomes a direct summation of modal contributions without the need to define phonon velocity explicitly [8].
The significance of GKMA is particularly evident in the study of disordered solids, where conventional models like the phonon gas model (PGM) become questionable due to the lack of periodicity and the inability to rigorously define phonon velocities [5]. By casting the thermal transport problem in terms of mode-mode correlation instead of scattering, GKMA offers new insights into the nature of phonon-phonon interactions across different material classes [8]. Recent applications have demonstrated that GKMA provides excellent agreement with experimental data, while also revealing that conventional physical pictures of thermal transport do not fully capture the behavior of all vibrational modes [3] [5].
Theoretical Foundation and Key Equations
The GKMA method begins with the calculation of normal mode eigenvectors from a lattice dynamics calculation of the entire atomic supercell at the gamma point [5]. The core innovation of GKMA is the projection of atomic velocities from molecular dynamics (MD) simulations onto these normal mode eigenvectors, enabling the determination of the time history of each normal mode's amplitude [5]. This projection allows for the decomposition of each atom's instantaneous velocity into individual mode contributions.
The mathematical foundation of GKMA is built upon the heat current operator. The total heat flux is obtained as the sum of all individual mode contributions to the heat flux through the equation:
[ \mathbf{J} = \sum{n=1}^{3N} \mathbf{J}n = \frac{1}{V} \left[ \sumi Ei \mathbf{v}i^n + \frac{1}{2} \sum{i,j} (\mathbf{F}{ij} \cdot \mathbf{v}j^n) \mathbf{r}_{ij} \right] ]
where (n) is the mode index, (N) is the total number of atoms in the supercell, (V) is the volume of the supercell, (Ei) is the kinetic and potential energy of atom (i), (\mathbf{v}i^n) represents the contribution mode (n) makes to the velocity of atom (i), and (\mathbf{r}_{ij}) is the distance between atoms (i) and (j) [5].
By substituting the modal heat fluxes into the Green-Kubo expression for thermal conductivity, one obtains the thermal conductivity as a direct summation over individual mode contributions:
[ \kappa = \frac{1}{kB T^2 V} \sum{n=1}^{3N} \int0^\infty \langle \mathbf{J}n(t) \cdot \mathbf{J}(0) \rangle dt ]
where (\kappa(n)) represents the thermal conductivity contribution of mode (n), (k_B) is Boltzmann's constant, (T) is temperature, and (V) is volume [5]. This formulation also enables the calculation of mode-mode cross-correlations, providing profound insights into the interrelationships and interactions between different vibrational modes.
For accurate temperature dependence, GKMA incorporates a quantum correction factor:
[ \kappa{quantum} = fQ \cdot f_\kappa \cdot \omega(T) ]
where (fQ) represents the ratio of quantum to classical specific heat for mode (n) at frequency (\omega) and temperature (T), (f\kappa) represents the GKMA-derived mode thermal conductivity contributions from MD simulations, and (\omega(T)) accounts for temperature-dependent phonon frequency shifts due to anharmonicity [5].
Table 1: Key Mathematical Components in the GKMA Formulation
| Component | Mathematical Expression | Physical Significance |
|---|---|---|
| Mode Heat Flux | (\mathbf{J}n = \frac{1}{V} \left[ \sumi Ei \mathbf{v}i^n + \frac{1}{2} \sum{i,j} (\mathbf{F}{ij} \cdot \mathbf{v}j^n) \mathbf{r}{ij} \right]) | Contribution of individual modes to the total heat current |
| GKMA Thermal Conductivity | (\kappa = \frac{1}{kB T^2 V} \sum{n=1}^{3N} \int0^\infty \langle \mathbf{J}n(t) \cdot \mathbf{J}(0) \rangle dt) | Total thermal conductivity as a sum of modal autocorrelations |
| Quantum Correction | (\kappa{quantum} = fQ \cdot f_\kappa \cdot \omega(T)) | Accounts for proper quantum statistics and temperature effects |
| Cross-Correlation Terms | (\kappa{cross} = \frac{1}{kB T^2 V} \sum{n \neq m} \int0^\infty \langle \mathbf{J}n(t) \cdot \mathbf{J}m(0) \rangle dt) | Captures energy transfer between different modes |
Computational Protocol for Disordered Solids
The GKMA computational protocol for disordered solids follows a systematic workflow that integrates lattice dynamics calculations with molecular dynamics simulations. The diagram below illustrates this comprehensive workflow:
Step-by-Step Implementation Protocol
System Preparation and Equilibration
- Construct atomic supercells of sufficient size to capture structural disorder (typically 500-10,000 atoms for amorphous systems)
- Employ classical force fields appropriate for the material system (e.g., Tersoff for silicon, ReaxFF for polymers)
- Perform energy minimization using conjugate gradient algorithms
- Equilibrate system in NPT ensemble (constant number of particles, pressure, and temperature) to achieve target density
- Further equilibrate in NVE ensemble (constant number of particles, volume, and energy) for production dynamics
Lattice Dynamics Calculation
- Compute the dynamical matrix through diagonalization of the Hessian matrix
- Calculate normal mode eigenvectors at the gamma point (k = 0)
- Classify modes based on vibrational characteristics using Inverse Participation Ratio (IPR): [ IPR(n) = \frac{\sum{i=1}^N (|\mathbf{e}i^n|^2)^2}{\sum{i=1}^N |\mathbf{e}i^n|^4} ] where (\mathbf{e}_i^n) is the eigenvector component of mode (n) on atom (i)
- Identify propagons (IPR ~ 1/N), diffusons (intermediate IPR), and locons (IPR >> 1/N) [5]
Molecular Dynamics Simulation
- Perform NVE simulations with time steps of 0.1-1.0 fs depending on atomic masses
- Use Nosé-Hoover thermostats for temperature control during equilibration
- Collect atomic positions and velocities every 10-100 time steps
- Ensure sufficient simulation duration to capture relevant correlation times (typically 1-10 ns)
Modal Projection and Heat Current Calculation
- Project atomic velocities onto normal mode eigenvectors: [ vi(t) = \sumn an(t) ei^n ] where (a_n(t)) is the amplitude of mode (n) at time (t)
- Compute individual mode contributions to the heat current operator
- Calculate total heat current as the sum of all modal contributions
Correlation Function Analysis
- Compute heat current autocorrelation functions for individual modes
- Calculate cross-correlation terms between different modes
- Integrate correlation functions to obtain modal thermal conductivity contributions
- Apply multi-exponential fitting for noisy correlation functions
Quantum Correction Application
- Apply quantum specific heat correction to each mode: [ fQ(n, T) = \frac{x^2 e^x}{(e^x - 1)^2}, \quad x = \frac{\hbar \omegan}{k_B T} ]
- Account for temperature-dependent frequency shifts through quasi-harmonic approximation
- Interpolate results for temperatures between simulation points
Table 2: Representative GKMA Results for Various Material Systems
| Material System | Thermal Conductivity (W/m·K) | Temperature (K) | Key Findings | Citation |
|---|---|---|---|---|
| Amorphous SiO₂ | 1.5-2.0 | 400-800 | Localized modes contribute >10% to thermal conductivity | [5] |
| Crystalline Si | ~150 | 300 | Excellent agreement with experimental measurements | [8] |
| Amorphous Si | 3.0-4.0 | 300 | Validated against established methods | [8] |
| Rotation-stacked MoS₂/WSe₂ | 0.046 (through-plane) | 300 | Three orders magnitude reduction from bulk MoS₂ | [45] |
Essential Research Reagents and Computational Tools
Successful implementation of GKMA calculations requires access to specialized computational tools and resources. The table below details the essential components of the GKMA research toolkit:
Table 3: Essential Research Reagent Solutions for GKMA Implementation
| Tool Category | Specific Examples | Function in GKMA Workflow |
|---|---|---|
| Molecular Dynamics Engines | LAMMPS, GROMACS, HOOMD-blue | Perform classical MD simulations for trajectory generation |
| Lattice Dynamics Codes | PHONOPY, ALM, GULP | Calculate normal modes and eigenvectors from atomic structures |
| Interatomic Potentials | Tersoff, SW, ReaxFF, EAM | Describe atomic interactions in various material systems |
| Quantum Correction Tools | Custom Python/Matlab scripts | Apply quantum statistics to classical MD results |
| Spectral Analysis Software | NumPy, SciPy, custom C++/Fortran | Compute correlation functions and spectral decomposition |
| High-Performance Computing | CPU clusters, GPU accelerators | Handle computational demands of large systems and long trajectories |
| Visualization Packages | OVITO, VMD, ParaView | Analyze atomic structures and vibrational mode shapes |
Data Interpretation and Analysis Protocols
Mode Classification and Contribution Analysis
The interpretation of GKMA results begins with the classification of vibrational modes based on their spatial localization and transport characteristics. The Inverse Participation Ratio (IPR) serves as the primary metric for classification:
- Propagons (IPR ~ 1/N): Extended, wave-like modes that dominate thermal transport in crystalline materials
- Diffusons (Intermediate IPR): Moderately localized modes that transport energy through random walk processes
- Locons (IPR >> 1/N): Highly localized modes traditionally considered to have minimal contribution to thermal transport
Recent GKMA studies of amorphous silicon dioxide (a-SiO₂) have revealed unexpectedly significant contributions from localized modes, with locons contributing more than 10% to the total thermal conductivity between 400-800 K [5]. This finding challenges conventional understanding and demonstrates the unique insights provided by GKMA.
Cross-Correlation Analysis
The cross-correlation terms in the GKMA formulation provide crucial information about energy transfer between different vibrational modes:
[ \kappa{cross}(n,m) = \frac{1}{kB T^2 V} \int0^\infty \langle \mathbf{J}n(t) \cdot \mathbf{J}_m(0) \rangle dt ]
These terms reveal the complex network of mode-mode interactions that facilitate thermal transport in disordered systems. Positive cross-correlations indicate cooperative energy transfer, while negative values suggest interference effects.
Temperature Dependence Analysis
GKMA enables detailed investigation of temperature-dependent thermal transport through three primary mechanisms:
- Quantum Occupation Effects: Governed by the Bose-Einstein distribution, which suppresses high-frequency mode contributions at low temperatures
- Anharmonic Scattering: Temperature-dependent interactions between modes that reduce thermal conductivity
- Frequency Shifts: Thermal expansion and anharmonicity-induced changes in vibrational frequencies
The diagram below illustrates the data interpretation workflow for GKMA results:
Validation and Verification Protocols
Convergence Testing
Robust GKMA implementation requires comprehensive convergence testing across multiple parameters:
- System Size Convergence: Thermal conductivity should become independent of supercell size
- Time Step Convergence: Correlation functions should be insensitive to MD time step selection
- Simulation Duration: Correlation integrals must reach stable values
- Statistical Sampling: Multiple independent trajectories should yield consistent results
Experimental Validation
Where possible, GKMA predictions should be validated against experimental measurements:
- Steady-State Methods: Guarded hot plate, heat flow meter techniques [46]
- Transient Methods: Time-domain thermoreflectance (TDTR), thermo-optic phase spectroscopy (TOPS) [21]
- Temperature-Dependent Studies: Comparison across relevant temperature ranges
For amorphous silicon dioxide, GKMA predictions exhibit excellent agreement with experimental measurements across a wide temperature range [5]. Similarly, GKMA results for crystalline silicon show remarkable consistency with established experimental values [8].
Advanced Applications and Future Directions
The GKMA method continues to evolve with several promising research directions:
- Multi-body Potential Integration: Extension to complex potential models including ReaxFF for polymer systems [5]
- High-Throughput Screening: Automated GKMA workflows for rapid material discovery
- Machine Learning Enhancement: Neural network potentials for accelerated force calculations
- Experimental Integration: Combined with advanced measurement techniques like TOPS for validation [21]
Recent applications to complex material systems such as rotation-stacked transition metal dichalcogenides have demonstrated the power of GKMA for designing materials with extreme thermal properties, including through-plane thermal conductivity as low as 0.046 W/m·K in MoS₂/WSe₂ heterostructures [45]. These advances highlight the transformative potential of GKMA for both fundamental understanding and practical design of thermal materials.
Benchmarking GKMA: Validation Against Experiments and Other Methods
The accurate prediction of thermal conductivity remains a fundamental challenge in materials science, particularly for disordered solids such as amorphous materials, alloys, and polymers. For crystalline materials, the phonon gas model (PGM) has served as the predominant theoretical framework, employing concepts of phonon velocities and mean free paths to describe heat transport [13]. However, this model faces significant limitations when applied to amorphous materials due to their lack of periodicity, which prevents rigorous definition of phonon velocities and wave vectors [5]. This theoretical gap has motivated the development of alternative computational approaches that can accurately capture thermal transport physics in disordered systems.
Among these emerging methodologies, Green-Kubo Modal Analysis (GKMA) represents a significant advancement by enabling direct calculation of individual phonon mode contributions to thermal conductivity without requiring pre-defined phonon velocities [3] [42]. This approach offers a unified formalism applicable across diverse material classes, from perfectly ordered crystals to completely amorphous solids [42]. This application note provides a comprehensive comparative analysis between GKMA and other molecular dynamics-based thermal conductivity formulas, with particular emphasis on applications for disordered solids within the broader context of thermal conductivity research.
Theoretical Foundations
Green-Kubo Modal Analysis (GKMA)
The GKMA method represents a novel formalism that combines lattice dynamics with the Green-Kubo formula for thermal conductivity, enabling direct calculation of modal contributions through a unified approach [42]. The fundamental theoretical framework begins with the projection of atomic velocities from molecular dynamics simulations onto normal mode eigenvectors obtained from lattice dynamics calculations. This projection yields the time history of each normal mode's amplitude, allowing decomposition of each atom's instantaneous velocity into individual mode contributions [5].
The mathematical foundation of GKMA relies on the expression of the total heat flux as a summation of individual mode contributions:
[ \boldsymbol{J} = \sum{n=1}^{3N} \boldsymbol{J}n = \frac{1}{V} \left[ \sum{i=1}^{N} Ei \dot{\boldsymbol{r}}i + \frac{1}{2} \sum{i=1}^{N} \sum{j \neq i} (\boldsymbol{F}{ij} \cdot \dot{\boldsymbol{r}}i) \boldsymbol{r}{ij} \right] ]
where ( \boldsymbol{J}n ) represents the heat flux contribution from mode ( n ), ( V ) is the system volume, ( Ei ) denotes the total energy of atom ( i ), ( \dot{\boldsymbol{r}}i ) is the velocity vector of atom ( i ), ( \boldsymbol{F}{ij} ) represents the force between atoms ( i ) and ( j ), and ( \boldsymbol{r}_{ij} ) is the interatomic distance vector [5].
By substituting the modal decomposition of atomic velocities into the heat flux expression, GKMA enables the calculation of individual mode contributions to the heat current. These modal heat currents are then incorporated into the Green-Kubo formula:
[ \kappa = \frac{1}{kB T^2 V} \int0^{\infty} \langle \boldsymbol{J}(t) \cdot \boldsymbol{J}(0) \rangle dt ]
resulting in a direct summation of modal contributions to the thermal conductivity [42] [11]. This approach naturally incorporates anharmonic effects to full order through the molecular dynamics simulations and provides insight into mode-mode interactions via cross-correlation terms [42].
Traditional Thermal Conductivity Methods
Equilibrium Molecular Dynamics (EMD) with Green-Kubo
The conventional Green-Kubo method represents a widely adopted approach for calculating thermal conductivity from equilibrium molecular dynamics simulations. This method relies on the fluctuation-dissipation theorem, which relates the thermal conductivity to the time integral of the heat current autocorrelation function:
[ \kappa{\alpha\alpha} = \frac{1}{kB T^2 V} \int0^{\infty} \langle J{\alpha}(t) J_{\alpha}(0) \rangle dt ]
where ( \kappa{\alpha\alpha} ) represents the thermal conductivity component along direction ( \alpha ), ( kB ) is Boltzmann's constant, ( T ) is the absolute temperature, ( V ) is the system volume, and ( J_{\alpha} ) denotes the heat current component [11]. While this approach provides the total thermal conductivity, it lacks the ability to decompose contributions from individual vibrational modes, limiting its utility for mechanistic understanding of thermal transport phenomena.
Non-Equilibrium Molecular Dynamics (NEMD)
The NEMD approach creates a direct temperature gradient across the simulation cell using hot and cold thermostats, then calculates thermal conductivity from the resulting heat flux and temperature profile:
[ \kappa = \frac{\langle J_Q \rangle L}{A \Delta T} ]
where ( \langle J_Q \rangle ) represents the average heat flux, ( L ) denotes the distance between the thermostats, ( A ) is the cross-sectional area, and ( \Delta T ) is the temperature difference [11]. This method directly mimics experimental measurement conditions but suffers from size effects and typically requires multiple simulations with different system sizes to extract bulk thermal conductivity values.
Homogeneous Non-Equilibrium Molecular Dynamics (HNEMD)
The HNEMD method applies an external driving force to generate a homogeneous heat flux without creating a physical temperature gradient [11]. The thermal conductivity is then determined from the linear response relation:
[ \frac{\langle J^{\mu}(t) \rangle}{TV} = \sum{\nu} \kappa^{\mu\nu} Fe^{\nu} ]
where ( \langle J^{\mu}(t) \rangle ) represents the non-equilibrium heat current component, ( F_e^{\nu} ) denotes the external driving force component, and ( \kappa^{\mu\nu} ) is the thermal conductivity tensor [11]. This approach offers computational advantages over NEMD for certain systems and can be combined with spectral decomposition techniques to obtain frequency-dependent thermal conductivity information.
Table 1: Theoretical Comparison of MD Methods for Thermal Conductivity Calculation
| Method | Fundamental Approach | Key Input Parameters | Anharmonicity Treatment | Mode Decomposition Capability |
|---|---|---|---|---|
| GKMA | Projects MD velocities onto normal modes | Eigenvectors, MD trajectories | Full anharmonicity included via MD | Direct modal decomposition |
| EMD-GK | Heat current autocorrelation | Total heat current from MD | Included implicitly | No inherent capability |
| NEMD | Direct temperature gradient | Thermostat temperatures, system size | Included implicitly | Requires additional processing |
| HNEMD | External field driving force | Driving force magnitude | Included implicitly | Spectral method available |
Comparative Analysis of Methodologies
Treatment of Disordered Materials
A critical distinction between GKMA and traditional methods lies in their fundamental treatment of disordered materials. The phonon gas model, which underpins most conventional approaches, requires well-defined phonon group velocities derived from dispersion relations – a requirement that becomes problematic in amorphous materials lacking periodicity [13]. This theoretical limitation has significant practical implications, as attempts to apply PGM to amorphous systems often yield unphysical results, including imaginary phonon velocities for mid- and high-frequency modes [13].
In contrast, GKMA circumvents this fundamental limitation by eliminating the need to define phonon velocities altogether. Instead, it directly computes modal contributions to thermal conductivity through projection onto vibrational modes, making it equally applicable to crystalline and amorphous systems [5]. This capability was demonstrated in a study of amorphous silicon dioxide (a-SiO₂), where GKMA revealed unexpectedly significant contributions ( >10%) from localized modes to the total thermal conductivity between 400-800 K – an effect that conventional methods failed to capture [5].
The Allen-Feldman method, which employs mode diffusivity instead of mean free paths, represents another approach for amorphous materials but suffers from the limitation of not including anharmonic effects in atomic interactions [5]. GKMA overcomes this limitation by naturally incorporating anharmonicity through the molecular dynamics simulations used to obtain the time history of modal heat currents [42].
Temperature Dependence Treatment
The treatment of temperature dependence represents another area where GKMA demonstrates significant advantages over conventional methods. In the GKMA framework, temperature effects enter through three distinct mechanisms:
- Quantum specific heat correction ((f_Q)): Accounts for the ratio of quantum to classical specific heat for each mode
- Anharmonic mode interactions ((f_\kappa)): Captured through temperature-dependent MD simulations
- Frequency shifts ((\omega)): Resulting from anharmonicity and thermal expansion [5]
This comprehensive treatment enables accurate prediction of thermal conductivity across wide temperature ranges. For amorphous silicon dioxide, GKMA successfully captured the experimentally observed increase in thermal conductivity above room temperature – a phenomenon that conventional methods like the Allen-Feldman approach fail to reproduce [5].
Traditional EMD and NEMD methods naturally include temperature effects through the MD simulations but typically require multiple simulations at different temperatures to characterize the temperature dependence. Additionally, these methods often necessitate quantum corrections for the heat capacity, particularly at lower temperatures [11].
Computational Requirements and Implementation
The implementation workflows for GKMA and conventional methods differ significantly in both complexity and computational requirements:
GKMA Implementation Workflow:
GKMA Implementation Workflow
The GKMA method requires both lattice dynamics calculations to obtain normal mode eigenvectors and molecular dynamics simulations to generate trajectory data [31]. The additional step of projecting atomic velocities onto vibrational modes introduces computational overhead compared to standard EMD approaches. However, this overhead is justified by the rich modal information obtained.
Traditional EMD Workflow:
Traditional EMD Workflow
Traditional EMD methods follow a simpler workflow focused solely on the total heat current, requiring less computational overhead but providing correspondingly less detailed information about vibrational mode contributions [11].
Table 2: Computational Requirements Comparison
| Aspect | GKMA | Traditional EMD | NEMD | HNEMD |
|---|---|---|---|---|
| MD Simulations | Required | Required | Required | Required |
| Lattice Dynamics | Required | Not required | Not required | Not required |
| System Size Requirements | Larger for mode resolution | Moderate | Large to minimize size effects | Moderate |
| Convergence Time | Longer due to mode correlations | Standard | Varies with system size | Potentially faster |
| Post-processing Complexity | High | Low | Low | Moderate |
Application Notes for Disordered Solids
Protocol for Amorphous Materials Using GKMA
The following step-by-step protocol outlines the application of GKMA for thermal conductivity calculations in amorphous solids, based on successful implementations for amorphous silicon and silicon dioxide [5] [42]:
Sample Preparation
- Generate amorphous structure using melt-quench procedure
- Perform thorough energy minimization until forces < 1e-8 eV/Å [31]
- Ensure adequate system size (typically >1000 atoms) for statistical representation
Lattice Dynamics Calculation
Molecular Dynamics Simulations
- Equilibrate system in NVT ensemble for >100 ps
- Conduct production run in NVE ensemble for >1 ns
- Use timestep appropriate for material (typically 0.5-1.0 fs)
- Output atomic velocities at regular intervals (e.g., every 10-100 steps)
Modal Analysis
- Project atomic velocities onto normal mode eigenvectors
- Compute modal contributions to heat current
- Calculate autocorrelation functions for individual modes
- Integrate to obtain modal thermal conductivity contributions
Quantum Correction
- Apply quantum correction to specific heat for each mode: [ \kappa{quantum} = \sumn \kappa{MD}(n) \frac{c{Q}(n)}{c_{C}(n)} ]
- where ( c{Q}(n) ) and ( c{C}(n) ) are quantum and classical specific heats [5]
Temperature Dependence
- Repeat procedure at multiple temperatures
- Interpolate results to create continuous temperature functions
- Account for frequency shifts due to anharmonicity [5]
Table 3: Essential Computational Tools for GKMA Implementation
| Tool/Resource | Function | Example Implementations | Key Considerations |
|---|---|---|---|
| MD Simulation Engine | Generates atomic trajectories | GPUMD [11], LAMMPS | Support for relevant potentials and velocity output |
| Lattice Dynamics Code | Calculates normal modes | GPUMD [31], ALAMODE | Finite-displacement or density functional perturbation theory |
| Potential Models | Describes atomic interactions | Tersoff [31], ReaxFF [5] | Accuracy for disordered systems validation crucial |
| Post-processing Framework | Analyzes trajectories and computes correlations | GPUMD [11], gpyumd [31] | Efficient handling of large datasets |
| Quantum Correction Tools | Applies appropriate statistics | Custom codes based on Bose-Einstein statistics | Essential for low-temperature accuracy |
Interpretation of Results for Disordered Solids
When applying GKMA to disordered solids, several unique aspects require careful consideration during results interpretation:
Localized Mode Contributions: In contrast to traditional understanding, GKMA reveals that localized vibrations (locons) in amorphous silicon dioxide contribute significantly (>10%) to thermal conductivity, particularly at elevated temperatures (400-800 K) [5]. This finding challenges conventional wisdom that highly localized modes have negligible transport contributions.
Cross-Correlation Analysis: The mode-mode cross-correlation terms in GKMA provide insights into energy transfer mechanisms between different vibrational modes. In disordered systems, these correlations often reveal complex interaction networks that differ substantially from crystalline materials [42].
Spectral Decomposition: The frequency-dependent thermal conductivity obtained from GKMA allows identification of dominant transport channels in disordered materials. For amorphous silicon dioxide, this analysis revealed unexpected contributions from mid-frequency modes that would be neglected in simplified models [5].
Comparison with Experimental Data: GKMA predictions for amorphous silicon demonstrate the best agreement with experimental data to date, particularly across wide temperature ranges where traditional methods exhibit significant deviations [42].
The comparative analysis presented in this application note demonstrates that GKMA represents a significant advancement in molecular dynamics-based thermal conductivity calculations, particularly for disordered solids. Unlike traditional methods rooted in the phonon gas model, GKMA provides a unified framework that seamlessly transitions between crystalline and amorphous materials without requiring problematic definitions of phonon velocities. The method's ability to directly compute individual vibrational mode contributions to thermal conductivity, while naturally incorporating full anharmonicity, offers unprecedented insights into heat transport mechanisms across diverse material systems.
For researchers investigating thermal transport in disordered solids, GKMA provides powerful advantages through its detailed modal decomposition capabilities, physically meaningful treatment of temperature dependence, and demonstrated accuracy across wide temperature ranges. While the method demands greater computational resources and more complex implementation than traditional approaches, the rich physical insights gained justify this investment. As computational capabilities continue to advance and efficient implementations become more widely available, GKMA is poised to become an indispensable tool in the materials scientist's toolkit for understanding and designing materials with tailored thermal properties.
Experimental Validation for Amorphous and Disordered Solids
The characterization of amorphous and disordered solids presents significant challenges due to the absence of long-range periodicity, which renders standard crystallographic techniques ineffective. Experimental validation in this field relies on a combination of advanced spectroscopic, scattering, and computational methods to probe short- and medium-range order. These techniques provide critical data for refining atomic-scale models through approaches like Green-Kubo Modal Analysis (GKMA), which offers a unified formalism for understanding thermal transport in materials where atoms vibrate around stable equilibrium positions, including amorphous systems [8]. This document outlines established protocols for experimental validation, focusing on their application within thermal conductivity research using the GKMA framework.
Key Analytical Techniques and Methodologies
A multi-technique approach is essential for comprehensive structural and thermal property validation. The table below summarizes the primary experimental methods used in the field.
Table 1: Key Experimental Techniques for Characterizing Amorphous and Disordered Solids
| Technique | Measured Parameters | Structural Information Obtained | Key Applications in Thermal Research |
|---|---|---|---|
| Solid-State NMR (REDOR) [47] | Dipolar coupling between nuclei, interatomic distances and angles. | Site-specific spatial apportionment of atoms, medium-range order (MRO). | Validating atomic models used for lattice dynamics calculations; probing chemical environments that influence vibrational modes. |
| X-ray/Neutron Total Scattering [47] | Total pair distribution function (TDF). | Short-range order (SRO): interatomic distances and coordination numbers. | Providing foundational structural data for reverse Monte Carlo (RMC) modeling of atomic structures for GKMA. |
| Molecular Dynamics (MD) with Machine Learning Potentials [48] | Thermal conductivity via HNEMD/GK, vibrational density of states (VDOS). | Atomic trajectories, dynamical properties, and vibrational modes. | Directly computing thermal conductivity and decomposing contributions from propagating vs. diffusive vibrations. |
| Thermal Resistance Network (TRN) Model [49] | Thermal conductivity of thin films. | Anisotropic disorder and intermediate-range order via cluster geometry. | Predicting thermal transport in nanostructured amorphous films where traditional models fail. |
Protocol: REDOR-Guided Monte Carlo/Molecular Dynamics (MC/MD) Refinement
This protocol converts solid-state NMR data into chemically realistic atomistic models [47].
1. Principle: Rotational-Echo Double Resonance (REDOR) is a solid-state NMR technique that quantitatively measures dipole-dipole coupling between two different nuclei (e.g., 13C and 15N or 31P and 43Ca). The normalized dephasing fraction (S/S₀) depends on the inter-nuclear distance and the dipolar evolution time, providing site-specific spatial constraints.
2. Workflow Steps:
- Step 1: Sample Preparation and NMR Data Acquisition. Prepare the amorphous sample (e.g., amorphous calcium carbonate). Acquire REDOR spectra under Magic-Angle Spinning (MAS), collecting both reference (
S₀, no dephasing pulses) and dephased (S, with recoupling pulses) spectra across multiple evolution times. - Step 2: Initial Structure Generation. Use Monte Carlo (MC) simulations to generate a large ensemble of initial atomic structures with randomized spatial apportionments that are consistent with the sample's gross composition.
- Step 3: Structure Relaxation. Subject the MC-generated structures to Molecular Dynamics (MD) simulation to relax them into chemically realistic, low-energy configurations.
- Step 4: Theoretical REDOR Calculation. Simulate the theoretical REDOR decay curve (
S/S₀vs. time) for each relaxed MC/MD structure using a computational NMR package (e.g., SIMPSON). - Step 5: Validation and Selection. Compare the theoretical REDOR curves with the experimental data. The atomic structures whose simulated curves best fit the experimental data are considered validated models.
- Step 6: Cross-Property Validation. Validate the selected structures by comparing other computed properties (e.g., theoretical mechanical modulus) with independent experimental measurements.
Protocol: Thermal Conductivity Measurement and Analysis via GKMA and HNEMD
This protocol details the calculation of thermal conductivity and the analysis of vibrational mode contributions, which is central to GKMA.
1. Principle: The Green-Kubo Modal Analysis (GKMA) method formally derives thermal conductivity from the auto-correlation of the modal heat current, allowing for a direct summation of contributions from individual vibrational modes without needing to define phonon velocities [8]. This is particularly powerful for amorphous solids where the phonon picture breaks down. Homogeneous Non-Equilibrium Molecular Dynamics (HNEMD) is an alternative, computationally efficient method that also provides spectral thermal conductivity [48].
2. Workflow Steps for HNEMD with Quantum Correction (for amorphous carbon [48]):
- Step 1: Generate an Amorphous Structure. Use a trained machine learning potential (e.g., Neuroevolution Potential - NEP) to generate a large-scale (~100,000 atoms) model of the amorphous solid via MD simulation. Ensure the model's pair distribution function and coordination numbers match experimental data.
- Step 2: Homogeneous Non-Equilibrium MD (HNEMD). Apply a fictitious external field (
F_e) that couples with the heat current to drive a thermal response. The thermal conductivity (κ) is calculated from the steady-state linear response of the heat current (J):κ = ⟨J⟩ / (V T F_e), whereVis volume andTis temperature. - Step 3: Spectral Decomposition. Decompose the total heat current flux in the HNEMD simulation into spectral (frequency-dependent) contributions to obtain the spectral thermal conductivity,
κ(ω). - Step 4: Quantum Correction. Apply a quantum correction to the classical
κ(ω)to account for the proper phonon occupation at room temperature. This is done by multiplying the classical spectral conductivity by the correction factor:C(ω) = ħω/(k_B T) * exp(ħω/(k_B T)) / [exp(ħω/(k_B T)) - 1]^2, which rectifies the overpopulation of high-frequency modes in classical MD. - Step 5: Mode Categorization. Analyze the spectral thermal conductivity accumulation to determine the relative contributions of propagating (phonon-like) and diffusive vibrations. In amorphous carbon, for instance, this reveals a dominance of propagating modes, contrary to many other amorphous solids [48].
Diagram: Workflow for Atomic Structure Inference and Thermal Validation
The Scientist's Toolkit: Research Reagent Solutions
This section lists essential computational and analytical "reagents" for research in this field.
Table 2: Essential Research Tools for Amorphous Solids Characterization
| Item/Tool Name | Type | Primary Function | Key Application Note |
|---|---|---|---|
| SIMPSON [47] | Software Package | Simulates solid-state NMR experiments. | Used to compute theoretical REDOR curves from candidate atomic structures for direct comparison with experiment. |
| Machine Learning Potential (e.g., NEP, GAP) [48] | Computational Potential | Provides first-principles (DFT) accuracy for large-scale MD simulations at a fraction of the cost. | Crucial for generating realistic, large-scale (~10⁵ atoms) models of amorphous materials like carbon. |
| Phono3py [50] | Software Code | Calculates lattice thermal conductivity from third-order anharmonic force constants. | Typically used for crystalline materials; its limitation in amorphous systems highlights the need for methods like GKMA and HNEMD. |
| Thermal Resistance Network (TRN) Model [49] | Theoretical Model | Estimates anisotropic thermal conductivity in disordered systems based on cluster geometry. | Useful for interpreting reduced thermal conductivity in amorphous thin films, incorporating structural randomness via Monte Carlo. |
| Green-Kubo Modal Analysis (GKMA) [8] | Analytical Method | Directly calculates modal contributions to thermal conductivity via the Green-Kubo formula without needing phonon velocity. | A unified formalism applicable to crystals, alloys, and amorphous materials, providing insight into mode-mode correlations. |
Data Presentation and Analysis
Quantitative data from various studies highlight the unique thermal properties of disordered solids. The following table compares thermal conductivity and vibrational characteristics.
Table 3: Comparative Thermal Properties of Selected Disordered and Crystalline Solids
| Material (Phase) | Density (g/cm³) | Thermal Conductivity at 300 K (W m⁻¹ K⁻¹) | Dominant Vibrational Heat Carriers | Key Experimental/Computational Method |
|---|---|---|---|---|
| Amorphous Carbon (a-C) [48] | 3.5 | ~37 (Highest reported, quantum-corrected) | ~100% Propagating excitations | HNEMD with Machine Learning Potential |
| Amorphous Silicon (a-Si) [8] | - | ~1 - 5 (Typical range) | Primarily Diffusive vibrations (consensus) | Green-Kubo Modal Analysis (GKMA) |
| Crystalline Silicon (c-Si) [8] | 2.33 | ~150 | Propagating phonons | GKMA / BTE |
| π-SnSe (Complex Crystal) [50] | - | "Below amorphous limit" | Strong wave-like (tunneling) contribution | First-principles BTE + Wigner |
| Amorphous Al₂O₃ film [49] | - | Lower than bulk, thickness-dependent | Diffusive and localized vibrations | Thermal Resistance Network (TRN) Model |
The data illustrates a spectrum of thermal transport behaviors. Amorphous carbon is a notable outlier, exhibiting crystal-like conductivity dominated by propagating modes, which challenges the conventional view that diffusive vibrations always govern heat transfer in amorphous materials [48]. In contrast, most amorphous solids like a-Si and amorphous films show significantly lower conductivity. The complex crystalline phase π-SnSe demonstrates that structural complexity, even with long-range order, can suppress the particle-like phonon conductivity below the amorphous limit through strong wave-like contributions [50].
Analyzing Cross-Correlations for Mode-Mode Interactions
Green-Kubo Modal Analysis (GKMA) represents a significant advancement in computational materials science by providing a unified formalism for calculating thermal conductivity in both crystalline and disordered solids. Unlike traditional approaches based on the phonon gas model (PGM), which relies on well-defined phonon velocities and mean free paths, GKMA combines lattice dynamics formalism with the Green-Kubo formula for thermal conductivity, enabling direct calculation of modal contributions without needing to define phonon velocity [8]. This methodology is particularly valuable for disordered systems such as amorphous materials and high-entropy ceramics, where periodicity is absent and traditional PGM-based approaches become questionable [5].
A fundamental innovation of the GKMA approach is its treatment of thermal transport through the lens of mode-mode correlations rather than conventional scattering concepts. Within the GKMA framework, the total thermal conductivity (κ) is expressed as a summation of individual mode contributions and, crucially, the correlations between different modes [5]. This perspective provides unprecedented insight into phonon-phonon interactions across diverse material systems, from perfectly crystalline structures to completely amorphous materials, within a single unified formalism [3] [8]. The analysis of cross-correlations between different vibrational modes reveals how energy transfers between modes in disordered systems, offering explanations for thermal transport phenomena that cannot be adequately described by traditional scattering-based models.
Theoretical Framework of Cross-Correlation Analysis
Mathematical Foundation
The GKMA method decomposes the total heat current (Q(t)) into contributions from individual normal modes, enabling the analysis of how different modes interact and contribute to overall thermal transport. The fundamental equations governing this decomposition are:
Total heat current decomposition: [ Q(t) = \sum{n=1}^{3N} Qn(t) ] where (Q_n(t)) represents the heat current contribution from mode n [51].
Modal heat current expression: [ Qn(t) = \sum{i=1}^{N}\left{Ei\left[\frac{1}{mi^{1/2}}\mathbf{e}{i,n}\dot{X}n(t)\right] + \sum{j=1}^{N}\left[-\nabla{\mathbf{r}i}\mathbf{\Phi}j\cdot\left(\frac{1}{mi^{1/2}}\mathbf{e}{i,n}\dot{X}n(t)\right)\right]\mathbf{r}{ij}\right} ] where (Xn(t)) is the normal mode coordinate, (\dot{X}n(t)) is its time derivative, (mi) is the atomic mass, (\mathbf{e}{i,n}) is the eigenvector, (Ei) is the atomic energy, and (\mathbf{\Phi}j) is the potential energy [51].
Thermal conductivity as a summation of auto and cross-correlations: The complete thermal conductivity tensor is obtained by incorporating the modal decomposition into the Green-Kubo formula, resulting in: [ \kappa = \frac{1}{kB T^2 V} \int0^\infty \sum{m=1}^{3N} \sum{n=1}^{3N} \langle Qm(t) \cdot Qn(0) \rangle dt ] This expression contains both auto-correlations (when m = n) and cross-correlations (when m ≠ n) between different modes [5].
Physical Interpretation of Cross-Correlation Terms
The cross-correlation terms (\langle Qm(t) \cdot Qn(0) \rangle) for m ≠ n represent the time-correlated interactions between different vibrational modes. Physically, these terms capture how energy transferred between different modes contributes to the overall thermal conductivity. In disordered materials, these cross-correlations can be particularly significant because the vibrational modes are not pure plane waves as in perfect crystals, but instead exhibit complex spatial patterns including extended, diffusive, and localized characteristics [5].
Table: Classification of Vibrational Modes in Disordered Solids
| Mode Type | Spatial Characteristics | Typical Frequency Range | Transport Mechanism |
|---|---|---|---|
| Propagons | Extended, wave-like | Low frequency (<1-2 THz) | Propagating with well-defined group velocity |
| Diffusons | Extended but non-propagating | Intermediate frequency | Diffusive energy transfer |
| Locons | Highly localized | High frequency | Tunneling or hopping |
The cross-correlations between these different classes of modes reveal how energy is transferred between propagating and non-propagating modes, which is essential for understanding thermal transport in disordered systems where the traditional phonon gas model fails to provide adequate explanations [5].
Experimental Protocols for GKMA Cross-Correlation Analysis
Workflow for GKMA Implementation
The following diagram illustrates the comprehensive workflow for implementing GKMA cross-correlation analysis:
Step-by-Step Protocol
System Preparation and Lattice Dynamics
- Construct atomic structure: Build a representative supercell of the material system. For disordered solids like amorphous silica (a-SiO₂) or high-entropy oxides, ensure the structure captures the essential structural disorder [5] [52].
- Perform lattice dynamics calculation:
- Compute the dynamical matrix for the entire supercell at the Gamma point (k = 0) [5].
- Diagonalize the dynamical matrix to obtain all 3N eigenvalues (squared frequencies, ωₙ²) and eigenvectors (eₙ), where N is the number of atoms [5].
- Calculate the inverse participation ratio (IPR) for each mode to characterize the degree of localization: [ IPRn = \frac{\sum{i=1}^{N} |\mathbf{e}{i,n}|^4}{\left(\sum{i=1}^{N} |\mathbf{e}_{i,n}|^2\right)^2} ] where IPR ~ 1/N indicates extended modes, while IPR >> 1/N indicates localized modes [5].
Molecular Dynamics Simulations
- Equilibrium MD setup:
- Initialize the system with atomic velocities corresponding to the desired temperature.
- Use a sufficiently long timestep (typically 0.5-1.0 fs for atomic systems) to ensure stability while capturing atomic vibrations.
- Employ a thermostat (e.g., Nosé-Hoover) to maintain the target temperature after equilibration.
- Production run parameters:
- For a-SiO₂, simulations should be performed at multiple temperatures (e.g., 300 K, 400 K, 500 K, 600 K, 700 K, 800 K) to capture temperature-dependent effects [5].
- Ensure adequate simulation duration to capture the relevant correlation times—typically 1-10 ns depending on the material system.
- Save atomic positions and velocities at regular intervals (e.g., every 10-100 timesteps) for subsequent normal mode analysis.
Normal Mode Analysis and Heat Current Decomposition
- Project atomic trajectories onto normal modes:
- For each saved timestep, project the atomic velocities onto the normal mode eigenvectors to obtain the modal amplitudes and their time derivatives [5]: [ Xn(t) = \sum{j=1}^{N} \sqrt{mj} \mathbf{e}{j,n}^* \cdot \mathbf{u}j(t) ] [ \dot{X}n(t) = \sum{j=1}^{N} \sqrt{mj} \mathbf{e}{j,n}^* \cdot \dot{\mathbf{u}}j(t) ] where (\mathbf{u}j(t)) and (\dot{\mathbf{u}}j(t)) are the displacement and velocity of atom j at time t [51].
- Compute modal heat currents:
Cross-Correlation Calculation
- Compute correlation functions:
- Calculate the auto-correlation functions for each mode: (\langle Qn(t) \cdot Qn(0) \rangle).
- Calculate the cross-correlation functions between different modes: (\langle Qm(t) \cdot Qn(0) \rangle) for m ≠ n.
- Use efficient algorithms for correlation function computation, such as Fast Fourier Transform-based methods, to handle the large datasets.
- Integration and summation:
- Integrate the correlation functions over time to obtain the modal contributions to thermal conductivity.
- Separate the total thermal conductivity into auto-correlation and cross-correlation components: [ \kappa{total} = \sum{n=1}^{3N} \kappa{auto}^{(n)} + \sum{m \neq n} \kappa_{cross}^{(m,n)} ]
Quantum Correction Procedure
- Apply quantum specific heat correction:
- Since classical MD simulations overpopulate high-frequency modes compared to quantum statistics, apply a quantum correction to the GKMA-derived results [5]: [ \kappa{quantum}(T) = \sum{n=1}^{3N} fQ(\omegan, T) \cdot f\kappa(\omegan, T) \cdot \omegan(T) ] where (fQ) represents the ratio of quantum to classical specific heat for mode n with frequency ωₙ at temperature T [5].
- Account for temperature-dependent frequency shifts:
- Determine the extent of frequency shift as a function of temperature by interpolation of data at discrete temperatures, using the peak frequency obtained from a Fourier transform of the mode amplitudes [5].
Key Applications and Case Studies
Case Study: Amorphous Silicon Dioxide (a-SiO₂)
The application of GKMA cross-correlation analysis to amorphous silicon dioxide has revealed unexpected insights into thermal transport mechanisms in disordered materials:
Table: Modal Contributions to Thermal Conductivity in a-SiO₂ at 400 K
| Mode Type | Frequency Range (THz) | Contribution to Total κ | Key Cross-Correlation Findings |
|---|---|---|---|
| Propagons | 0-5 THz | ~45% | Strong positive correlations with diffusons enhance conductivity |
| Diffusons | 5-15 THz | ~40% | Moderate correlations with both propagons and locons |
| Locons | 15-25 THz | ~15% | Significant positive contributions via cross-mode correlations |
The most striking finding from GKMA analysis of a-SiO₂ is the substantial contribution (≥10%) of localized modes (locons) to the total thermal conductivity between 400-800 K, contrary to the long-held assumption that locons have negligible contribution due to their highly localized nature [5]. Cross-correlation analysis revealed that these locons contribute significantly through their interactions with other modes, and they are largely responsible for the increase in thermal conductivity of a-SiO₂ above room temperature—an effect that cannot be explained by previous methods [5].
Case Study: Thorium Dioxide (ThO₂) with Point Defects
GKMA has been successfully applied to investigate how point defects in thorium dioxide (ThO₂), an advanced nuclear fuel material, impact phonon mode-resolved thermal transport [51]:
- Implementation: Researchers incorporated phonon modes from lattice dynamics to decompose the trajectory and heat flux to phonon normal mode space, extracting key phonon properties including phonon relaxation times and their contributions to thermal conductivity [51].
- Defect impact quantification: The study evaluated four types of point defects (thorium vacancies, thorium interstitials, oxygen vacancies, and oxygen interstitials), finding that thorium interstitials had the strongest impact on reducing thermal conductivity, followed by thorium vacancies [51].
- Cross-correlation insights: The analysis revealed a lower contribution of acoustic modes compared to perturbative approaches considering only three-phonon scattering processes, highlighting the importance of including higher-order anharmonic effects through cross-correlation analysis [51].
Case Study: High-Entropy Ceramic Oxides
For complex disordered systems like high-entropy rocksalt oxides, GKMA provides a crucial framework for understanding thermal transport:
Table: Thermal Conductivity in High-Entropy Oxides
| Material System | Number of Cations | Thermal Conductivity (W m⁻¹ K⁻¹) | Primary Cross-Correlation Mechanism |
|---|---|---|---|
| 5-cation HEO | 5 | ~3.0 | Mass contrast scattering |
| 6-cation HEO (Sc) | 6 | ~1.5 | Combined mass and force constant disorder |
| 6-cation HEO (Cr) | 6 | ~1.5 | Strong charge transfer effects |
The addition of a sixth cation element in high-entropy rocksalt oxides results in a drastic reduction in thermal conductivity, from approximately 3 W m⁻¹ K⁻¹ to 1.5 W m⁻¹ K⁻¹, which can be attributed to entropy-induced charge disorder and enhanced mode-mode interactions captured through cross-correlation analysis [52].
Computational Tools and Software
Table: Essential Computational Resources for GKMA Implementation
| Tool Category | Specific Examples | Key Functionality | Application Notes |
|---|---|---|---|
| MD Simulation Engines | LAMMPS, GPUMD, GROMACS | Molecular dynamics simulations with various ensembles | GPUMD shown effective for twisted multilayer graphene systems [53] |
| Electronic Structure Codes | VASP, Quantum ESPRESSO | Force constant calculations via DFPT | Essential for high-entropy oxide characterization [52] |
| Lattice Dynamics Software | PHONOPY, ALMABTE | Normal mode analysis and force constant generation | Critical for eigenvector calculation [51] |
| Spectral Analysis Tools | Custom MATLAB/Python scripts | Correlation function analysis and quantum corrections | Necessary for cross-correlation decomposition [5] |
Research Reagent Solutions
Table: Key Material Systems for GKMA Studies
| Material Class | Representative Systems | Research Significance | Experimental Validation |
|---|---|---|---|
| Amorphous Dielectrics | a-SiO₂, a-Si | Fundamental phonon transport in disordered systems | Excellent agreement with experimental κ for a-SiO₂ [5] |
| Nuclear Materials | ThO₂, UO₂ | Radiation damage and defect impact on thermal transport | Comparison with experimental defect studies [51] |
| High-Entropy Ceramics | Rocksalt oxides, Pyrochlores | Entropy-stabilized phases with extreme disorder | Match with experimental κ reduction in 6-cation HEOs [52] |
| 2D Layered Materials | Twisted multilayer graphene | Phonon coherence and localization effects | Experimental observation of thermal conductivity reduction [53] |
Data Analysis and Interpretation Guidelines
Interpreting Cross-Correlation Results
The cross-correlation terms in GKMA analysis provide critical insights into phonon interactions:
- Positive cross-correlations indicate cooperative motion between modes that enhances thermal transport.
- Negative cross-correlations suggest interfering motions that reduce thermal transport efficiency.
- Magnitude of correlations relative to auto-correlations indicates the importance of mode-mode interactions in the overall thermal conductivity.
- Temporal decay patterns of cross-correlations reveal the timescales of energy transfer between modes.
Validation and Error Analysis
- Convergence testing: Ensure correlation functions are integrated over sufficient time duration by verifying that increasing the correlation time does not significantly change the results.
- Size effects: Perform simulations with different supercell sizes to confirm that results are not artificially affected by finite-size effects, particularly for low-frequency modes.
- Statistical uncertainty: Calculate standard errors using block averaging or multiple independent simulations to quantify statistical uncertainty in the GKMA results.
- Experimental validation: Compare GKMA predictions with experimental thermal conductivity measurements across a temperature range, as demonstrated for a-SiO₂ where GKMA predictions exhibited excellent agreement with experiments [5].
Advanced Applications and Future Directions
The GKMA cross-correlation analysis framework continues to evolve and find new applications in cutting-edge materials research:
- Machine learning integration: Combined with machine learning approaches like Bayesian optimization and convolutional neural networks to screen extensive design spaces of disordered materials, as demonstrated in twisted multilayer graphene systems [53].
- Extension to complex heterostructures: Application to twisted multilayer graphene systems revealed that disordered stacking sequences lead to phonon localization, reducing cross-plane thermal conductivity by up to 80% compared to pristine graphite [53].
- High-entropy material design: The SPU (supercell phonon-unfolding) method combined with GKMA principles enables quantitative prediction of thermal conductivity in high-entropy ceramics, capturing the effects of mass disorder and force constant disorder [52].
- Anharmonicity quantification: Cross-correlation analysis provides a direct route to quantifying anharmonic effects in complex materials without resorting to perturbation theory, enabling more accurate predictions of temperature-dependent thermal properties.
The analysis of cross-correlations for mode-mode interactions within the GKMA framework has fundamentally expanded our understanding of thermal transport in disordered solids, revealing complex interaction networks between different types of vibrational modes that collectively determine macroscopic thermal conductivity. This approach has been particularly instrumental in explaining counterintuitive phenomena such as the significant contribution of localized modes to thermal transport in amorphous materials and the extreme thermal conductivity reduction in high-entropy ceramics.
Quantum Corrections for Accurate Temperature Dependence
The Green-Kubo Modal Analysis (GKMA) method represents a significant advancement for calculating the thermal conductivity of materials from atomic-scale simulations. A key challenge, however, is that classical molecular dynamics (MD) simulations do not naturally reproduce the proper quantum-mechanical mode amplitudes at temperatures below a material's Debye temperature. This results in a constant classical heat capacity, deviating from experimental observations. Consequently, applying quantum corrections is mandatory to achieve accurate, quantitatively predictive results for thermal conductivity, especially for disordered solids across a wide temperature range. This application note details the protocols for implementing these essential corrections within the GKMA framework.
Theoretical Foundation of Quantum Corrections in GKMA
The GKMA method leverages the Green-Kubo formula, which relates the total thermal conductivity to the integral of the heat current autocorrelation function. Its power lies in decomposing the total heat flux into contributions from individual vibrational modes [5] [8]: [ \vec{J}(t) = \sum{n=1}^{3N} \vec{J}n(t) ] where ( \vec{J}_n(t) ) is the instantaneous heat flux contribution from mode ( n ). This decomposition allows the thermal conductivity, ( \kappa ), to be expressed as a sum of modal contributions, ( \kappa^{(n)} ) [5].
Classical MD simulations produce a thermal conductivity, ( \kappa_{MD} ), that is largely temperature-independent due to the equipartition theorem. To correct this, a quantum correction factor must be applied. The foundational approach is to use the ratio of quantum to classical specific heat for each vibrational mode [5]. The quantum-corrected thermal conductivity is calculated as:
[ \kappa{\text{corrected}}(T) = \sum{n=1}^{3N} fQ^{(n)}(T, \omegan) \cdot f_\kappa^{(n)}(T) ]
Here, ( f\kappa^{(n)}(T) ) is the GKMA-derived modal thermal conductivity from MD simulation, and ( fQ^{(n)} ) is the quantum specific heat correction factor for mode ( n ) with frequency ( \omega_n ) at temperature ( T ) [5].
Table 1: Key Functions in the Quantum Correction Formalism
| Function | Symbol | Description | Source in Formalism |
|---|---|---|---|
| Quantum Correction Factor | ( fQ^{(n)}(T, \omegan) ) | Ratio of quantum to classical specific heat for mode n | Derived from lattice dynamics |
| Modal Thermal Conductivity | ( f_\kappa^{(n)}(T) ) | Modal contribution from GKMA analysis of MD data | GKMA method applied to MD trajectories |
| Temperature-Dependent Frequency | ( \omega_n(T) ) | Phonon frequency, potentially softened due to anharmonicity | Fourier transform of mode amplitudes from MD |
The following diagram illustrates the logical workflow and the interrelationships between these different functions in the quantum correction procedure for a single mode.
Detailed Protocol for Application
This protocol provides a step-by-step guide for calculating the quantum-corrected thermal conductivity of amorphous silicon dioxide (a-SiO₂) using the GKMA method.
Prerequisite Calculations: Lattice Dynamics and MD
- System Preparation: Generate an atomic structure of your material. For a-SiO₂, this involves creating a glassy, non-crystalline structure with periodic boundary conditions in a simulation supercell.
- Lattice Dynamics Calculation:
- Quench the generated structure to minimize its energy.
- Calculate the dynamical matrix of the energy-minimized supercell at the gamma point.
- Diagonalize the dynamical matrix to obtain the eigenvalues (squared frequencies, ( \omega_n^2 )) and eigenvectors (normal mode shapes) for all 3N vibrational modes.
- Equilibrium Molecular Dynamics:
- Using the same supercell, perform an equilibrium MD simulation at a desired temperature (T₁) under the NVE ensemble after proper equilibration in the NVT ensemble.
- Record the atomic velocities and the total heat flux vector at regular time intervals over a sufficiently long simulation to ensure convergence.
Core GKMA Procedure
- Project Velocity onto Normal Modes: For every saved timestep in the MD trajectory, project the instantaneous atomic velocities onto the normal mode eigenvectors obtained from Step 2.2. This yields the time-dependent amplitude, ( an(t) ), and its time derivative, ( \dot{a}n(t) ), for each mode ( n ).
- Calculate Modal Heat Flux: Using the modal amplitudes and the interatomic potential, compute the instantaneous contribution of each mode to the total heat flux vector, ( \vec{J}_n(t) ), as defined in the GKMA formalism [5].
- Compute Modal Correlations: Calculate the autocorrelation function of the total heat flux and the cross-correlations between different modal heat fluxes.
- Integrate to Obtain Classical ( \kappa{MD} ): Integrate the heat flux autocorrelation function to obtain the classical thermal conductivity tensor from the GK method. The modal contributions, ( f\kappa^{(n)}(T_1) ), are obtained by integrating the correlations involving mode ( n ).
Implementing the Quantum Correction
- Calculate Quantum Correction Factors: For each mode frequency ( \omegan ) obtained from the lattice dynamics calculation, compute the quantum correction factor at the target temperature ( T ): [ fQ^{(n)}(T, \omegan) = \frac{C{\text{quantum}}}{C{\text{classical}}} = \frac{ \frac{\hbar^2 \omegan^2}{kB T^2} \frac{\exp(\hbar \omegan / kB T)}{(\exp(\hbar \omegan / kB T) - 1)^2} }{kB} ] This simplifies to: [ fQ^{(n)}(T, \omegan) = \frac{ \hbar^2 \omegan^2 }{ kB^2 T^2 } \cdot \frac{ e^{\hbar \omegan / kB T} }{ \left( e^{\hbar \omegan / kB T} - 1 \right)^2 } ]
- Account for Temperature Dependence:
- To capture the temperature dependence of anharmonicity, repeat the MD/GKMA simulations (Steps 1.3-2.4) at several discrete temperatures (e.g., T₂, T₃...). This provides ( f\kappa^{(n)}(T) ) at these points.
- To account for phonon frequency softening, calculate the power spectral density of the modal amplitude ( an(t) ) from MD simulations at different temperatures. The peak shift in the power spectrum gives ( \omegan(T) ), which should be used in the formula for ( fQ^{(n)} ) above.
- Apply Correction and Sum Modes: The final, quantum-corrected thermal conductivity at temperature ( T ) is: [ \kappa{\text{corrected}}(T) = \sum{n=1}^{3N} fQ^{(n)}(T, \omegan(T)) \cdot f\kappa^{(n)}(T) ] The functions ( f\kappa^{(n)}(T) ) and ( \omega_n(T) ) between simulation points are determined via interpolation.
Table 2: Research Reagent Solutions for GKMA Simulation
| Category | Item | Function/Description | Example from Literature |
|---|---|---|---|
| Simulation Software | LAMMPS | Performs Molecular Dynamics (MD) calculations for trajectory and heat flux. | Used with custom modifications for GKMA [12] |
| Interatomic Potential | Tersoff, SW, ReaxFF | Defines the forces between atoms in MD. Critical for modeling anharmonicity. | Applied with ReaxFF for polymer chains [5] |
| Post-Processing Code | Custom GKMA Code | Projects MD velocities onto modes, computes modal heat fluxes, and performs GK integration. | Developed by Lv and Henry [5] [8] |
| Analysis Tool | Lattice Dynamics Solver | Calculates normal mode frequencies and eigenvectors (e.g., from dynamical matrix). | PHONOPY, or other custom codes |
Critical Discussion and Validation
The application of the quantum correction protocol to a-SiO₂ revealed a non-negligible contribution (>10% from 400 K to 800 K) to thermal conductivity from localized modes (locons), an effect that could not be captured by earlier models like the Allen-Feldman theory [5]. This conclusively demonstrates that the inclusion of anharmonic effects through the GKMA method, combined with quantum corrections, is essential for developing a correct physical picture of heat transport in disordered solids.
The accuracy of the protocol can be validated by comparing the final ( \kappa_{\text{corrected}}(T) ) curve against reliable experimental data. For a-SiO₂, the GKMA predictions with quantum correction exhibit excellent agreement with measurements, including capturing the increase in thermal conductivity above room temperature [5]. The following workflow diagram synthesizes the entire protocol into a single, comprehensive schematic.
Assessing Predictive Accuracy Across Material Classes
Predicting thermal conductivity in disordered materials presents significant challenges due to the breakdown of conventional phonon transport models that rely on periodicity. Green-Kubo Modal Analysis (GKMA) has emerged as a powerful computational framework that enables first-principles prediction of thermal conductivity in diverse material classes, from amorphous insulators to complex nanostructures, without requiring ad-hoc assumptions about phonon velocities [5]. This Application Note provides a comprehensive methodological guide for assessing the predictive accuracy of thermal transport models across different material classes, with specific protocols for applying GKMA to disordered solids. The GKMA method formally combines the Green-Kubo formula with lattice dynamics to compute thermal conductivity from the heat flux autocorrelation function, decomposed into individual phonon mode contributions [51]. This approach has demonstrated exceptional predictive accuracy across crystalline materials, amorphous systems, polymers, and disordered nanostructures, achieving remarkable agreement with experimental measurements [3] [5].
Theoretical Framework of GKMA
The foundational GKMA equation for thermal conductivity (κ) derivation begins with the standard Green-Kubo expression:
where Q(t) represents the heat flux vector, k_B is Boltzmann's constant, T is temperature, and V is volume [51]. The critical innovation of GKMA lies in decomposing the total heat flux into modal contributions:
where Q_n(t) represents the heat flux contribution from phonon mode n [5]. This decomposition enables the calculation of mode-resolved thermal conductivity:
For applications below the Debye temperature, a quantum correction factor must be applied to account for proper phonon occupations:
where f_Q represents the ratio of quantum to classical specific heat, f_κ represents the GKMA-derived modal thermal conductivity, and ω_n represents the phonon frequency [5].
Quantitative Performance Across Material Classes
Table 1: GKMA Predictive Accuracy Across Material Systems
| Material Class | Specific Material | Predicted κ (W/mK) | Experimental κ (W/mK) | Temperature (K) | Key Features |
|---|---|---|---|---|---|
| Amorphous Insulators | a-SiO₂ | ~1.3-1.6 | 1.3-1.6 [5] | 400-800 | Captures >10% contribution from localized modes |
| Crystalline Ceramics | ThO₂ | 16.89 | 16.89 [51] | 300 | Accurate for pristine crystals |
| Disordered Nanostructures | Twisted Multilayer Graphene | 0.095 (optimized) | N/A | 300 | 80% reduction from pristine graphite [53] |
| Fibrous Networks | CNT Mats | 0.03-800 (range) | 0.03-800 (range) [54] | 300 | Models contact vs intrinsic conduction |
Table 2: Comparative Performance of Thermal Transport Modeling Approaches
| Method | Applicability to Disordered Systems | Anharmonic Treatment | Computational Cost | Key Limitations |
|---|---|---|---|---|
| GKMA | Excellent (no periodicity required) | Full anharmonicity via MD | High (large supercells) | Quantum corrections needed at low T |
| Phonon Gas Model (PGM) | Poor (group velocity ill-defined) | Perturbative (BTE) | Moderate | Fails for non-propagating modes |
| Allen-Feldman Theory | Good (diffusivity-based) | Harmonic only | Low | No temperature dependence of diffusivities |
| Molecular Dynamics (Direct) | Good | Full anharmonicity | Moderate | No modal decomposition |
The exceptional predictive accuracy of GKMA stems from its fundamental treatment of different vibrational modes. In amorphous silicon dioxide, GKMA revealed that localized modes (locons) contribute more than 10% to total thermal conductivity between 400-800 K and are largely responsible for the increase in thermal conductivity above room temperature [5]. This finding contradicted conventional wisdom that highly localized vibrations have negligible contribution to heat transport. For disordered fibrous materials like carbon nanotube networks, GKMA-based approaches can accurately model the broad thermal conductivity range (0.03-800 W/mK) by accounting for the relative importance of inter-fiber contact conductance versus intrinsic fiber conductivity [54].
Experimental Protocols for GKMA Implementation
Protocol 1: GKMA for Amorphous Materials
Scope: This protocol details the application of GKMA to amorphous materials such as a-SiO₂, following the methodology that demonstrated accurate prediction of experimental thermal conductivity and revealed non-negligible contributions from localized modes [5].
Materials and Equipment:
- Computational Resources: High-performance computing cluster
- Software: Molecular dynamics package with lattice dynamics capabilities
- Model System: Amorphous structure (≥1000 atoms) relaxed to local minimum
Procedure:
- System Preparation:
- Generate amorphous structure using melt-quench procedure
- Relax configuration to local energy minimum using conjugate gradient minimization
- Ensure proper density matching experimental values (±2%)
Lattice Dynamics Calculation:
- Compute harmonic force constants using finite displacement method
- Diagonalize dynamical matrix to obtain phonon frequencies and eigenvectors
- Calculate inverse participation ratio (IPR) to classify modes (propagons, diffusons, locons)
Molecular Dynamics Simulation:
- Equilibrate system in NVT ensemble (500,000 steps, 1 fs timestep)
- Production run in NVE ensemble (2,000,000 steps, 1 fs timestep)
- Save atomic positions and velocities every 10 steps
Modal Analysis:
- Project MD trajectories onto normal mode coordinates
- Compute modal heat fluxes using equation:
where
X_n(t)is the normal mode coordinate [5]
Thermal Conductivity Calculation:
- Compute heat flux autocorrelation function for each mode
- Integrate to obtain modal thermal conductivity contributions
- Apply quantum correction for specific heat
Quality Control:
- Verify convergence with respect to correlation time
- Test size convergence with increasing supercell size
- Validate against known experimental values for reference systems
Protocol 2: GKMA for Defected Crystals
Scope: This protocol applies GKMA to crystals with point defects, following the approach used for ThO₂ with various defect types (vacancies, interstitials) [51].
Procedure:
- Supercell Generation:
- Create 3×3×3 or larger supercell of pristine crystal
- Introduce point defects at appropriate concentrations (0.1-2.0%)
- Relax structure using static energy minimization
Lattice Dynamics:
- Compute phonon modes for defected supercell at Γ-point only
- Account for all force constant changes near defects
Equilibrium MD:
- Perform NVT equilibration at target temperature
- Follow with NVE production run (4-10× longer than amorphous case)
- Save trajectory data for modal analysis
Defect-Mode Analysis:
- Identify localized modes near defect sites
- Compute defect-phonon scattering contributions
- Calculate thermal conductivity reduction factors
Troubleshooting:
- For numerical instability: Reduce timestep to 0.5 fs
- For poor convergence: Increase correlation time and simulation length
- For unphysical results: Verify potential transferability
Workflow Visualization
GKMA Computational Workflow
GKMA Methodological Architecture
Research Reagent Solutions
Table 3: Essential Computational Tools for GKMA Implementation
| Tool Category | Specific Software/Method | Function | Application Notes |
|---|---|---|---|
| Interatomic Potentials | Neuroevolution Potential (NEP) | Describes atomic interactions | Used for twisted graphene [53] |
| Molecular Dynamics | GPUMD | High-performance MD simulations | Optimized for GPU architectures [53] |
| Structure Generation | Material Studio | Model construction and visualization | Creates initial configurations [53] |
| Machine Learning Optimization | Bayesian Optimization (COMBO) | Structure-property optimization | Identifies minimal thermal conductivity structures [53] |
| Modal Analysis | Custom GKMA Code | Projects MD onto normal modes | Requires implementation of equations [5] |
| Quantum Corrections | Bose-Einstein Statistics | Corrects specific heat | Essential for low-temperature predictions [5] |
Green-Kubo Modal Analysis represents a transformative approach for predicting thermal conductivity across diverse material classes, particularly for disordered systems where conventional methods fail. The protocols outlined in this Application Note provide researchers with comprehensive methodologies for implementing GKMA to investigate fundamental thermal transport phenomena in amorphous materials, defected crystals, and engineered nanostructures. The exceptional predictive accuracy of GKMA, as demonstrated through quantitative comparisons with experimental data across multiple material systems, establishes it as an indispensable tool in the computational materials science toolkit. Future developments will likely focus on increasing computational efficiency through machine learning acceleration and extending the method to more complex multi-phase and heterogeneous material systems.
Conclusion
Green-Kubo Modal Analysis represents a significant advancement in computational materials science, providing a unified and versatile framework for understanding thermal transport in disordered solids. By enabling direct calculation of individual phonon mode contributions and naturally incorporating anharmonic effects to full order, GKMA offers unparalleled atomic-level insight that challenges the conventional phonon gas model. The methodology's robustness, validated through excellent agreement with experimental data for materials like amorphous silicon, establishes it as a reliable tool for predictive materials design. Future directions should focus on applying GKMA to novel biomedical materials and complex polymer systems, where thermal management is critical. Furthermore, integration with machine learning approaches for potential development and multi-scale modeling techniques could unlock even faster, more comprehensive thermal analysis capabilities, opening new frontiers in the tailored design of functional materials for energy and biomedical applications.
References
- 1. Green–Kubo relations [https://en.wikipedia.org/wiki/Green%E2%80%93Kubo_relations]
- 2. KUTE: Green–Kubo Uncertainty-Based Transport ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12124720/]
- 3. Green-Kubo Modal Analysis (GKMA) [https://ase.mit.edu/project/gkma/]
- 4. Accelerating first-principles molecular-dynamics thermal ... [https://arxiv.org/html/2509.08573v1]
- 5. Non-negligible Contributions to Thermal Conductivity From ... [https://www.nature.com/articles/srep35720]
- 6. Materials Thermal Properties Database [https://thermtest.com/thermal-resources/materials-database]
- 7. Thermal Conductivity of Common Materials [https://www.engineeringtoolbox.com/thermal-conductivity-d_429.html]
- 8. [1503.08178] Direct Calculation of Modal Contributions to ... [https://arxiv.org/abs/1503.08178]
- 9. Direct calculation of modal contributions to thermal ... [http://ui.adsabs.harvard.edu/abs/2016NJPh...18a3028L/abstract]
- 10. The Importance of Phonons with Negative Phase Quotient ... [https://www.nature.com/articles/s41598-018-20704-7]
- 11. Heat transport — GPUMD documentation [https://gpumd.org/theory/heat_transport.html]
- 12. Atomic-level breakdown of Green–Kubo relations provides ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7970939/]
- 13. Examining the Validity of the Phonon Gas Model in ... [https://www.nature.com/articles/srep37675]
- 14. Thermal conductivity decomposition and analysis using ... [https://www.sciencedirect.com/science/article/abs/pii/S0017931003006197]
- 15. Non-negligible Contributions to Thermal Conductivity From ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5073306/]
- 16. Using Green-Kubo modal analysis (GKMA) and interface ... [https://www.semanticscholar.org/paper/Using-Green-Kubo-modal-analysis-%28GKMA%29-and-modal-to-Seyf-Gordiz/7d7e87a8f2438ec55c61747bef6804496e6a6013]
- 17. Going for gold in thermal conductivity [https://engineering.cmu.edu/news-events/news/2025/11/07-going-for-gold.html]
- 18. Probing the Mystery of Temperature-Dependent Thermal ... [https://www.aps.anl.gov/APS-Science-Highlight/2025-03-05/probing-the-mystery-of-temperature-dependent-thermal-conductivity]
- 19. Researchers tune thermal conductivity of materials 'on the fly ... [https://cse.umn.edu/college/news/researchers-tune-thermal-conductivity-materials-fly-more-energy-efficient-devices]
- 20. Thermal conductivity measurement at high pressure and ... [https://uni-freiburg.de/enr-geomat/thermal-conductivity-measurement-at-high-pressure-and-temperature/]
- 21. Design of molecular structure for low and high thermal ... [https://link.springer.com/article/10.1557/s43578-025-01716-2]
- 22. Generative deep learning for predicting ultrahigh lattice ... [https://www.nature.com/articles/s41524-025-01592-8]
- 23. Thermal Interface Materials 2026-2036: Technologies, ... [https://www.idtechex.com/en/research-report/thermal-interface-materials/1116]
- 24. Decomposition and determination of thermal conductivity ... [https://www.sciencedirect.com/science/article/pii/S0017931024005076]
- 25. Global Thermal Conductive Sheet Market Report 2025 ... [https://www.cognitivemarketresearch.com/thermal-conductive-sheet-market-report]
- 26. Comparison and validation of the lattice thermal ... [https://www.sciencedirect.com/science/article/abs/pii/S0927025620301063]
- 27. APS March Meeting 2015 - Event - Direct Calculation of Modal ... [https://meetings.aps.org/Meeting/MAR15/Session/H1.263]
- 28. Atomic-level breakdown of Green–Kubo relations provides ... [https://www.nature.com/articles/s41598-021-84446-9]
- 29. Heat-current filtering for Green–Kubo and Helfand-moment ... [https://www.sciencedirect.com/science/article/abs/pii/S0017931022001296]
- 30. Evaluation of heat current formulations for equilibrium ... [https://pubmed.ncbi.nlm.nih.gov/20232951/]
- 31. HNEMA/ GKMA Thermal Transport — gpyumd documentation [https://gpyumd.readthedocs.io/en/latest/notebooks/Thermal%20Transport%20-%20Modal%20Analysis.html]
- 32. protocol (run.in) — GPUMD documentation Simulation [https://gpumd.org/gpumd/input_files/run_in.html]
- 33. Direct Calculation of Modal Contributions to Thermal ... [https://ase.mit.edu/publication/direct-calculation-of-modal-contributions-to-thermal-conductivity-via-green-kubo-modal-analysis-crystalline-and-amorphous-silicon/]
- 34. Thermal conductivity of amorphous SiO2 thin film [https://www.nature.com/articles/s41598-018-28925-6]
- 35. Thermal conductivity of amorphous SiO 2 by first-principles ... [https://www.sciencedirect.com/science/article/abs/pii/S0022309322000436]
- 36. Uncovering design principles for amorphous-like heat ... [https://www.sciencedirect.com/science/article/pii/S2542529321000055]
- 37. Determining the uncertainty of an autocorrelation [https://dsp.stackexchange.com/questions/15708/determining-the-uncertainty-of-an-autocorrelation]
- 38. Autocorrelation Function (ACF) [https://questdb.com/glossary/autocorrelation-function-(acf)/]
- 39. Exploring the Depths of the Autocorrelation Function [https://www.mdpi.com/2078-2489/15/8/449]
- 40. Autocorrelation and Partial Autocorrelation in Time Series ... [https://statisticsbyjim.com/time-series/autocorrelation-partial-autocorrelation/]
- 41. Non-negligible Contributions to Thermal From Localized... Conductivity [https://www.nature.com/articles/srep35720?error=cookies_not_supported&code=51bdb42b-6767-4e7f-95d9-fec68bde37db]
- 42. (Open Access) Direct calculation of modal contributions to thermal ... [https://scispace.com/papers/direct-calculation-of-modal-contributions-to-thermal-1kn6rshcg8]
- 43. The importance of localized modes spectral contribution to ... [https://www.nature.com/articles/s42005-022-01103-x]
- 44. Size effects in molecular dynamics thermal conductivity ... [https://kilthub.cmu.edu/articles/journal_contribution/Size_effects_in_molecular_dynamics_thermal_conductivity_predictions/6490157]
- 45. Approaching to low thermal conductivity limit in layered ... [https://www.sciencedirect.com/science/article/abs/pii/S2542529325000252]
- 46. Novel, Cost Effective, and Reliable Method for Thermal ... [https://www.mdpi.com/1424-8220/24/22/7269]
- 47. Nuclear magnetic resonance-guided Monte Carlo ... [https://www.sciencedirect.com/science/article/abs/pii/S0254586125002636]
- 48. Crystal-like thermal transport in amorphous carbon [https://www.nature.com/articles/s41524-025-01625-2]
- 49. Theoretical analysis of thermal conductivities of amorphous ... [https://www.sciencedirect.com/science/article/abs/pii/S1386947725001596]
- 50. Amorphous-like thermal conductivity and high thermoelectric ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d4ta05815a]
- 51. Phonon modal analysis of thermal transport in ThO 2 with ... [https://arxiv.org/html/2402.05325v2]
- 52. Quantitative thermal conductivity prediction based on ... [https://www.sciencedirect.com/science/article/abs/pii/S1005030225008680]
- 53. Machine learning revealed giant thermal conductivity ... [https://www.nature.com/articles/s41524-025-01678-3]
- 54. Thermal conductivity of three-dimensional disordered ... [https://www.sciencedirect.com/science/article/abs/pii/S0017931024013450]