Evaluating Coordination Number Determination Methods: Accuracy, Applications, and Future Directions for Biomedical Research
Accurately determining atomic coordination numbers is fundamental to understanding structure-property relationships in chemistry and materials science, with profound implications for drug development and biomedicine.
Evaluating Coordination Number Determination Methods: Accuracy, Applications, and Future Directions for Biomedical Research
Abstract
Accurately determining atomic coordination numbers is fundamental to understanding structure-property relationships in chemistry and materials science, with profound implications for drug development and biomedicine. This article provides a comprehensive evaluation of modern coordination number determination methods, from foundational geometric approaches to advanced techniques based on electron density and machine learning. We explore the principles, applications, and limitations of methods including Voronoi-Dirichlet partitioning, topological coordination analysis, X-ray absorption spectroscopy, and continuous symmetry measures. By comparing accuracy across different chemical environments and material systems, this review serves as an essential guide for researchers selecting appropriate characterization strategies for complex molecular systems, single-atom catalysts, and metalloproteins relevant to pharmaceutical development.
Fundamental Concepts and Challenges in Coordination Number Determination
The coordination number serves as a fundamental descriptor in materials science, chemistry, and physics, quantifying how many nearest neighbors surround a central atom or ion. While conceptually simple as a counting rule, practical determination reveals significant complexity, with multiple experimental and computational methodologies yielding divergent values from the same underlying structure. This comparison guide objectively evaluates the performance of leading coordination number determination methods, examining their underlying principles, accuracy limitations, and appropriate application domains. By synthesizing experimental benchmarks and computational validations, we provide researchers with a structured framework for selecting methodologies that align with their accuracy requirements and system characteristics, particularly focusing on challenges in liquid systems and nanomaterial characterization where traditional counting rules prove inadequate.
The coordination number represents a cornerstone structural parameter across scientific disciplines, informing understanding of packing efficiency, chemical bonding, and physical properties in systems ranging from simple crystals to complex amorphous materials and biological assemblies. Traditional definitions rely on radial cutoff distances from the central atom, yet this simplistic approach falters for noncrystalline systems where bond length distributions overlap and lack clear minima. The practical determination of coordination numbers reveals substantial methodological divergence, where identical systems yield different numerical values depending on the experimental or computational technique employed.
This methodological variability stems from fundamental differences in what physical property each technique actually probes. X-ray and neutron scattering methods measure pairwise correlation functions, while spectroscopic techniques probe electronic environments, and computational approaches rely on geometric or electronic structure analysis. Consequently, the "true" coordination number becomes method-dependent, necessitating a nuanced understanding of each technique's inherent assumptions, limitations, and accuracy boundaries. This guide systematically compares these approaches within a rigorous accuracy assessment framework, providing experimental validation data and practical implementation protocols to guide method selection for specific research applications.
Methodological Approaches and Experimental Protocols
Experimental Determination Methods
Scattering Techniques
Scattering methods derive coordination numbers through integration of the radial distribution function (RDF), which describes particle density as a function of distance from a reference atom. The coordination number between species α and β is calculated as:
Experimental Protocol for Neutron Scattering of Liquid Argon:
- Sample Preparation: Utilize high-purity argon (≥99.99%) sealed in sample cells with neutron-transparent windows (e.g., aluminum, vanadium). Maintain temperature control (±0.1 K) using cryostat systems.
- Data Collection: Employ time-of-flight neutron diffractometers with incident wavelengths typically 0.5-2.0 Å. Collect data at multiple scattering angles to achieve wide Q-range (0.1 to 30 Å⁻¹). Measurement duration typically 6-24 hours depending on flux and sample size.
- Data Processing: Subtract background scattering and container contributions. Apply multiple scattering corrections and normalize to absolute units using standard samples.
- Coordination Number Calculation: Fourier transform structure factor S(Q) to obtain g(r). For liquid argon at 85 K, integrate first coordination shell from Rmin = 3.10 Å to Rmax = 5.03 Å, where R_max corresponds to the first minimum in g(r) [1]. Use number density ρ₀ = 0.02125 atoms/ų for calculation.
Accuracy Considerations: The upper integration limit R* significantly impacts calculated coordination numbers. For liquid argon, empirical determination establishes R* = 5.03 Å at 85 K based on neutron scattering data [1]. Different integration methods yield coordination numbers ranging from 10.0 to 12.5 for identical datasets, highlighting methodological sensitivity.
Spectroscopic Approaches
Spectroscopic methods, particularly nuclear magnetic resonance (NMR), provide complementary coordination information through measurement of parameters sensitive to local atomic environment.
NMR Experimental Protocol for Organic Molecules:
- Sample Preparation: Dissolve target molecules in deuterated solvents at concentrations 10-100 mM. Add reference standards (e.g., TMS for ¹H/¹³C referencing) and ensure sample homogeneity.
- Data Collection: Acquire ¹H and ¹³C NMR spectra at controlled temperature (typically 298 K) using high-field spectrometers (≥400 MHz). For scalar coupling constants, utilize 2D experiments such as HMBC (nJCH) and COSY (nJHH) with sufficient digital resolution.
- Parameter Extraction: Measure proton-carbon (nJCH) and proton-proton (nJHH) scalar coupling constants through peak analysis. Validate experimental values against DFT-calculated benchmarks to identify misassignments [2].
- Structure Determination: Utilize measured NMR parameters as constraints for 3D structure determination through molecular modeling or DFT-based structure optimization.
Computational Determination Methods
Computational approaches offer atomic-level resolution but vary in their definition of "coordination" based on geometric or electronic criteria.
Geometric-Based Protocol:
- Cutoff Method: Identify neighbors within fixed distance threshold (e.g., first minimum of RDF). Simple but fails for disordered systems with broad bond length distributions.
- Voronoi Tessellation: Partition space into polyhedral cells around each atom. Coordination number equals number of faces, providing natural neighbor definition without distance cutoffs.
- Delaunay Triangulation: Construct tetrahedral network connecting atomic centers, useful for liquid structure analysis [1].
Electronic Structure Protocol:
- Population Analysis: Calculate coordination from molecular orbital occupancy or bond order analysis (e.g., Wiberg bond indices).
- DFT Calculations: Employ functionals such as mPW1PW91 with basis sets like 6-311 g(dp) to compute NMR parameters for validation against experimental data [2].
Comparative Performance Analysis
Method Accuracy Assessment
Table 1: Coordination Number Determination Method Comparison
| Method | Spatial Resolution | Accuracy Limit | System Applicability | Key Limitations |
|---|---|---|---|---|
| Neutron Scattering | Ensemble average | ±0.5 coordination | Liquids, amorphous materials | Integration limit sensitivity, isotopic requirements |
| X-ray Diffraction | Ensemble average | ±0.7 coordination | Crystalline solids, dense liquids | Limited light element sensitivity, radiation damage |
| NMR Spectroscopy | Atomic local environment | Coordination trends | Molecular systems, complexes | Indirect measurement, interpretation complexity |
| Voronoi Analysis | Atomic resolution | ±0.1 coordination | Computational models, packed systems | Boundary effects in low-density systems |
| Radial Distribution | Atomic resolution | ±0.3 coordination | All atomistic systems | Cutoff distance ambiguity, thermal broadening |
Table 2: Experimental Coordination Numbers for Liquid Argon (85 K) by Method
| Determination Method | Integration/Calculation Approach | Reported Coordination Number | Reference System |
|---|---|---|---|
| Neutron Scattering | RDF integration to R* = 5.03 Å | 10.0 | Yarnell et al. [1] |
| Neutron Scattering | RDF integration to first minimum | 12.5 | Mikolaj and Pings [1] |
| Voronoi Analysis | Face counting without cutoff | 13.8-14.2 | Computational models [1] |
| Theoretical Model | Barker et al. potential | 11.3 | DFT/MD simulations [1] |
| Theoretical Model | Bae potential | 12.1 | DFT/MD simulations [1] |
Temperature Dependence and System Effects
Coordination numbers exhibit significant temperature dependence, particularly in liquid systems. For liquid argon, coordination values decrease from approximately 12 near the triple point (83.81 K) to as low as 5 approaching the critical point (150.70 K) [1]. This substantial structural variation under different thermal conditions highlights the importance of reporting temperature alongside coordination number values and selecting methods appropriate for the system's thermodynamic state.
Different computational potentials yield varying coordination numbers, with the Bae potential showing results comparable to more complex, three-parameter models [1]. This sensitivity to potential selection underscores the need for careful method validation against experimental data when available.
Research Reagent Solutions Toolkit
Table 3: Essential Materials and Computational Resources
| Research Reagent/Resource | Function in Coordination Analysis | Application Notes |
|---|---|---|
| High-Purity Argon (≥99.99%) | Reference sample for scattering studies | Essential for calibration of liquid structure analysis |
| Deuterated NMR Solvents | Matrix for molecular structure determination | Enables high-resolution NMR parameter measurement |
| Neutron-Transparent Cells | Sample containment for scattering experiments | Aluminum/vanadium construction minimizes background |
| DFT Software (mPW1PW91) | Computational validation of experimental data | Benchmarking level for NMR parameter calculation [2] |
| LSH Forest Indexing | Efficient neighbor identification in large datasets | Enables analysis of datasets up to 10⁷ points [3] |
| TMAP Algorithm | Visualization of high-dimensional data relationships | Tree-based representation preserves neighborhood structure [3] |
Integrated Workflow for Accurate Determination
The determination of coordination numbers extends far beyond simple atom counting, encompassing diverse methodologies with inherent strengths, limitations, and accuracy boundaries. This comparative analysis demonstrates that methodological selection significantly impacts reported coordination numbers, with variations exceeding 20% observed across techniques applied to identical systems. Scattering methods provide ensemble averages with well-characterized precision but suffer from integration limit sensitivity. Spectroscopic techniques offer local environmental insight but require careful interpretation and computational validation. Geometric computational approaches deliver atomic-resolution data but depend on definitional criteria.
For researchers pursuing accurate coordination number determination, a multi-method approach with cross-validation provides the most reliable pathway. Specifically, computational studies should validate against experimental benchmarks where available, while experimental approaches should explicitly report integration limits, temperature conditions, and methodological assumptions. The development of standardized validation datasets, such as the NMR parameter collection for organic molecules [2], enables more rigorous method benchmarking. As structural characterization advances toward increasingly complex systems, acknowledging and quantifying the methodological dependence of coordination numbers becomes essential for accurate structural interpretation and material property prediction.
Historical Evolution from Geometric to Electronic Descriptors
The evolution from geometric to electronic descriptors represents a fundamental paradigm shift in computational chemistry and materials science. This transition marks a move from describing molecular structures based solely on their spatial arrangement to characterizing them by their electronic properties, which more directly govern chemical reactivity and function. Geometric descriptors, which condense the complex spatial arrangement of atoms into simplified numerical representations or counts, provided the foundational language for early structural chemistry [4]. For decades, the coordination number (CN) stood as a cornerstone geometric descriptor, traditionally defined simply as the number of atoms in the first sphere around a central atom [4]. However, the limitations of this purely geometric perspective became increasingly apparent, particularly for complex systems like intermetallic compounds or transition metal complexes, where chemical bonding mechanisms are not always clear-cut [4]. This spurred the development of electronic descriptors, which leverage the principles of quantum mechanics to describe the electron density distribution within a molecule, offering a more direct link to a material's chemical and catalytic properties [5] [6]. This guide objectively compares the accuracy, applications, and methodological underpinnings of these two descriptor classes within the specific context of evaluating coordination number determination methods.
Comparative Analysis of Descriptor Methodologies
The following tables provide a structured comparison of the fundamental characteristics and experimental validation of geometric and electronic descriptors.
Table 1: Fundamental Characteristics of Geometric and Electronic Descriptors
| Feature | Geometric Descriptors | Electronic Descriptors |
|---|---|---|
| Theoretical Basis | Classical geometry & graph theory [6] | Quantum mechanics & density functional theory [5] [6] |
| Primary Information | Nuclear coordinates, interatomic distances, connectivity [4] [6] | Electron density distribution, orbital energies [5] [6] |
| Key Examples | Topological Coordination Number (tCN), Voronoi-Dirichlet Polyhedra, Wiener Index [4] [6] | d-band model, Principal Component (PC) descriptors of density of states, Hirshfeld Charges, HOMO/LUMO energies [5] [6] [7] |
| Data Requirement | Crystal structure metrics (atomic coordinates) [4] | Computed electronic structure (e.g., from DFT calculations) [5] [7] |
| Interpretability | Intuitive but often lacks direct link to properties [6] | More abstract but offers direct connection to chemical activity [5] |
Table 2: Experimental Protocol and Performance Comparison
| Aspect | Geometric Descriptors | Electronic Descriptors |
|---|---|---|
| Primary Experimental Validation | X-ray crystallography, comparison with crystallographic databases (e.g., CSD) [4] [7] | X-ray Absorption Spectroscopy (XAS), optical spectroscopy, correlation with catalytic activity/chemisorption energy [5] [8] |
| Computational Workflow | Distance analysis (e.g., Brunner-Schwarzenbach), Voronoi tessellation, solid angle calculation [4] | Density Functional Theory (DFT) optimization, electronic structure analysis (e.g., using PCA) [5] [7] |
| Typical Workflow | Structure → Interatomic distances → Coordination number [4] | Structure → DFT Optimization → Electronic Structure Analysis → Descriptor [5] [7] |
| Handling of Dynamic/Disordered Systems | Problematic; relies on static snapshots [4] | More robust; AIMD simulations can model dynamic coordination fluctuations [8] |
| Reported Accuracy in Coordination Analysis | Can lack coordination reciprocity; may over-count neighbors (Voronoi) [4] | PCA-derived electronic descriptors show competitive accuracy in predicting chemisorption properties [5] |
Evolution of Experimental Protocols
Classic Geometric Determination Protocols
Traditional geometric approaches for determining coordination numbers rely heavily on crystallographic data. The Brunner-Schwarzenbach (BS) method is a classic protocol that identifies the first coordination sphere by locating the first significant gap in the sequence of increasing interatomic distances from a central atom [4]. A more advanced protocol involves Voronoi-Dirichlet Partitioning (VDP), which divides space into polyhedra, each containing all points closer to a given atom than to any other. The number of polyhedral faces gives the maximum coordination number, which is often refined using solid angles subtended by each face to produce a weighted, effective coordination number [4]. These methods, while powerful, face challenges such as a lack of coordination reciprocity and ambiguity in defining the exact boundary of the coordination sphere [4].
Modern Electronic Structure Analysis Protocols
Modern protocols for electronic descriptor analysis are rooted in quantum mechanical calculations. A standard workflow begins with Density Functional Theory (DFT) geometry optimization of the molecular or solid-state structure, often using functionals like B3LYP and basis sets such as 6-31G* [7]. Subsequently, the electronic structure is analyzed. One powerful protocol uses Principal Component Analysis (PCA) of the electronic density of states (DOS) [5]. In this unsupervised machine learning approach, the complex DOS data is decomposed into principal components that capture the most significant variations. These PC scores then serve as highly accurate, interpretable descriptors for predicting properties like chemisorption strength [5]. For analyzing complex, multi-component systems like metal ions in molten salts, a correlative protocol combining X-ray Absorption Spectroscopy (XAS), optical spectroscopy, and multivariate curve resolution (MCR-ALS) is employed. This protocol deconvolutes the spectral data to identify and quantify the population distribution of different coordination states coexisting in dynamic equilibrium [8].
Diagram 1: Workflow comparison of geometric and electronic descriptor generation and application.
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Software and Computational Tools for Descriptor Research
| Tool Name | Type | Primary Function in Descriptor Analysis |
|---|---|---|
| Gaussian | Software Suite | Performing DFT calculations for geometry optimization and electronic property derivation [7]. |
| RDKit | Open-Source Cheminformatics | Generating molecular fingerprints (e.g., Morgan fingerprints) and traditional 2D descriptors [9] [7]. |
| Amber | Molecular Simulation | Utilizing force field parameters (e.g., GAFF2) for molecular mechanics simulations [7]. |
| molSimplify | Open-Source Code | High-throughput generation and screening of transition metal complex structures [10]. |
| Demeter | Software Package | Processing and analyzing X-ray Absorption Spectroscopy (XAS) data [8]. |
| Cambridge Structural Database (CSD) | Database | Repository of experimental crystallographic data for validation and trend analysis [10] [7]. |
Accuracy Assessment in Coordination Number Determination
The accuracy of coordination number determination is highly context-dependent. Geometric descriptors, particularly the topological coordination number (tCN) based on the quantum theory of atoms in molecules (QTAIM), offer an improvement over traditional methods by naturally accounting for different atomic sizes and providing coordination-consistent scenarios [4]. However, they still face challenges in systems with high structural disorder or dynamic fluctuation.
Electronic descriptors, particularly those derived from machine learning analysis of electronic structure, demonstrate superior accuracy for predicting chemical activity. Studies show that principal-component (PC) descriptors of the electronic density of states yield machine learning models with competitive accuracy in predicting chemisorption properties on metal alloys and oxides compared to models using established descriptors [5]. Furthermore, in complex systems like molten salts, electronic structure analysis combined with AIMD simulations is essential for accurately identifying the population distribution of coordination states that dynamically coexist, a feat difficult to achieve with static geometric methods [8].
For drug discovery applications, the choice of descriptor impacts predictive performance. A comparative study on ADME-Tox targets found that models using traditional 1D, 2D, and 3D descriptors could outperform those based solely on molecular fingerprints for certain machine learning algorithms [9]. This underscores that the "best" descriptor is often task-dependent, but electronic and higher-dimensional descriptors generally provide a more chemically-grounded foundation for predicting biologically-relevant properties.
Diagram 2: Experimental protocol for electronic analysis of coordination states in complex systems like molten salts [8].
The historical evolution from geometric to electronic descriptors reflects the chemical sciences' progression from descriptive to predictive capabilities. While geometric descriptors remain invaluable for structural classification and initial analysis, their limitations in explaining and predicting chemical behavior are evident. Electronic descriptors, powered by advances in quantum mechanical computations and machine learning, provide a more fundamental and accurate link to a material's properties, from catalytic activity to speciation in complex fluids. The ongoing integration of these descriptor types, alongside sophisticated experimental validation protocols, continues to enhance the accuracy of coordination number determination and beyond, paving the way for the accelerated design of novel materials and drugs.
The coordination number (CN) is a fundamental descriptor in crystallography and materials science, condensing the complex spatial arrangement of atoms around a central atom into a single integer. Despite its conceptual simplicity, a significant challenge exists in moving beyond a purely geometric definition to one that is chemically meaningful and physically consistent. The determination of an accurate CN is often complicated by two principal factors: the requirement for coordination reciprocity and the influence of atomic size effects. This guide objectively compares the performance of established and emerging methods for CN determination, with a specific focus on how they address these two challenges, providing researchers with a framework for selecting the appropriate analytical tool.
The traditional approach of analyzing interatomic distances, such as the Brunner-Schwarzenbach method, often fails to deliver reciprocity, meaning that for the same A–B distance, atom A might be counted in the coordination sphere of B, but not vice versa. This violates physical chemistry principles, as bonding interactions are inherently pairwise symmetric [4]. Furthermore, methods based on Voronoi-Dirichlet partitioning (VDP), while providing a unique geometric CN, typically do not naturally account for differences in atomic sizes, requiring additional corrections and weighting schemes [4]. This guide evaluates methods based on their foundational principles, summarizing key differentiators in the table below.
Table 1: Comparison of Coordination Number Determination Methods
| Method | Fundamental Principle | Handles Coordination Reciprocity? | Accounts for Atomic Size Effects? | Primary Application Context |
|---|---|---|---|---|
| Distance-Based (e.g., Brunner-Schwarzenbach) | Identification of large gaps in interatomic distance sequences [4] | No (Often violates reciprocity) [4] | No (Purely metric) [4] | Traditional crystallographic analysis |
| Voronoi-Dirichlet Partitioning (VDP) | Geometric partitioning of space into atomic domains; atoms sharing a polyhedron face are neighbors [4] | Yes (By construction) | No (Requires empirical weighting e.g., solid angles) [4] | Intermetallic compounds, complex alloys [4] |
| Topological Coordination Number (tCN) | Analysis of electron density via QTAIM; solid angles of interatomic surfaces [4] | Yes (Includes reciprocity requirements) [4] | Yes (Naturally includes atomic electron-density decay) [4] | Quantum crystallography, intermetallic phases [4] |
| Data-Driven/Machine Learning (e.g., TOSS, GNNs) | Statistical learning from large datasets of known structures (e.g., ICSD, CSD) [11] [10] | Implicitly learned from data | Implicitly learned from data (e.g., via coordination radii) [11] | High-throughput materials screening, predicting novel complexes [11] [10] |
| Atomic Density Field | Coarse-grained field variable capturing local atomic packing [12] | Not a direct CN method (Provides a related descriptor) | Yes (Captures both neighbor count and interatomic spacing) [12] | Mesoscale modeling of defects (e.g., grain boundaries) [12] |
Experimental Protocols for Method Validation
The accuracy of any CN determination method must be validated against reliable experimental or computational benchmarks. The following protocols detail key methodologies used to generate reference data in this field.
In Silico Benchmarking with Ab Initio Molecular Dynamics (AIMD)
Objective: To compute reference structural properties, including coordination numbers, for phases under extreme or complex conditions where traditional experiments are challenging [13].
Workflow:
- System Setup: Construct a supercell of the crystal structure (e.g., 2000 atoms for BCC-Fe) to ensure thermodynamic stability and avoid finite-size effects [13].
- DFT Calculations: Perform AIMD simulations using software like VASP. Employ a plane-wave basis set with a defined energy cut-off (e.g., 400 eV) and the GGA for exchange-correlation energy. Use the NVT ensemble to maintain target temperature and pressure conditions [13].
- Data Collection: Run the simulation for thousands of timesteps (e.g., 8000 with a 1.0 fs timestep) to ensure proper equilibration.
- Structure Analysis: Compute the Radial Distribution Function (RDF) from the trajectory. The coordination number is derived by integrating the RDF up to the first minimum [13].
Supporting Experimental Data: This approach was crucial in resolving the structure of iron under Earth's core conditions. AIMD simulations of a 2000-atom BCC-Fe supercell at 560 GPa and ~7000 K produced a coordination number of ~11, successfully matching experimental EXAFS data that was previously ambiguous [13].
Multi-Modal Speciation in Molten Salts
Objective: To identify and quantify the population distribution of different coordination states of a metal ion (e.g., Ni(II)) in a molten salt matrix as a function of temperature and composition [14].
Workflow:
- Sample Preparation: Prepare molten salt mixtures (e.g., LiCl–KCl, NaCl–MgCl₂) with a low concentration (e.g., 1 wt%) of the metal salt (e.g., NiCl₂) in an inert atmosphere glovebox to prevent oxidation and moisture contamination [14].
- In Situ Spectroscopy:
- Data Deconvolution:
- Use Principal Component Analysis (PCA) on the optical spectra to determine the number of unique coordination states.
- Apply Multivariate Curve Resolution – Alternating Least Squares (MCR-ALS) analysis to the XANES data to extract pure spectral components for each coordination state and their mixing fractions [14].
- Structure Identification: The extracted spectral components are assigned to specific coordination geometries (e.g., tetrahedral, octahedral) by comparing them with spectra from AIMD simulations of candidate structures [14].
Data-Driven Oxidation State and Coordination Analysis
Objective: To determine chemically intuitive oxidation states (OS) and infer coordination environments in crystal structures from large datasets, providing a validation source for CN analysis [11].
Workflow (Tsinghua Oxidation States in Solids - TOSS):
- Data Ingestion: Preprocess a large dataset of crystal structures (e.g., from the Materials Project).
- Library Building ("Digesting"): For each atomic site, analyze its local coordination environment across a range of distance tolerance parameters. This process is repeated over the entire dataset to build a library of distance distributions and coordination radii for each element pair [11].
- OS Assignment ("Practicing"): For each structure, determine the OSs by minimizing a loss function based on the Bayesian maximum a posteriori probability (MAP), using the previously built library as the prior. The algorithm seeks the most probable OS assignment that is consistent with the observed local coordination environments across the dataset [11].
- Model Training: The resulting OS-labeled data can be used to train a Graph Convolutional Network (GCN) model for rapid OS prediction, achieving high accuracy (>97%) against human-curated labels [11].
Diagram 1: The TOSS workflow for data-driven oxidation state assignment.
The Scientist's Toolkit: Key Reagents and Computational Solutions
Table 2: Essential Reagents and Resources for Coordination Environment Research
| Reagent / Resource | Function / Description | Application Example |
|---|---|---|
| Cambridge Structural Database (CSD) | A repository of experimentally determined organic and metal-organic crystal structures used for data mining and trend analysis [10]. | Curating datasets of ligands with known metal-coordination behavior to train machine learning models [10]. |
| Vienna Ab initio Simulation Package (VASP) | A software package for performing DFT calculations and AIMD simulations [13]. | Modeling the structure and coordination of phases under extreme pressures and temperatures [13]. |
| molSimplify | An open-source software for the automated generation and analysis of transition metal complexes [10]. | High-throughput in silico structure generation and screening of TMCs with realistic coordination geometries [10]. |
| Graph Neural Networks (GNNs) | A class of machine learning models that operate on graph-structured data, ideal for molecular structures [10]. | Predicting the coordinating atoms and denticity of a ligand directly from its SMILES string [10]. |
| Multivariate Curve Resolution (MCR-ALS) | A chemometric analysis technique that deconvolutes mixed spectroscopic signals into pure components [14]. | Resolving the XANES spectra of Ni(II) in molten salts into contributions from distinct coordination states (e.g., 4-, 5-, 6-coordinate) [14]. |
The accurate determination of coordination numbers is a non-trivial problem at the heart of materials science and chemistry. As this guide demonstrates, the choice of method directly impacts the ability to resolve the core challenges of coordination reciprocity and atomic size effects. While traditional geometric methods are computationally simple, they often fail to deliver chemically consistent results. The emerging paradigm, exemplified by the topological CN approach and data-driven machine learning models, integrates physical principles with statistical learning to provide a more robust and meaningful analysis of atomic coordination. This progression is critical for developing reliable structure-property relationships, ultimately accelerating the design of new materials and catalysts.
The Critical Distinction Between Geometric and Chemical Coordination Numbers
In the fields of crystallography, inorganic chemistry, and materials science, the term "coordination number" (CN) is a fundamental descriptor of atomic architecture. However, its interpretation varies significantly depending on the conceptual framework applied, leading to a critical distinction between geometric coordination numbers and chemical coordination numbers. The geometric coordination number is a purely structural metric derived from the immediate spatial arrangement of atoms, calculated by counting neighboring atoms within a defined radial cutoff or by analyzing the geometry of coordination polyhedra [15] [16]. In contrast, the chemical coordination number is intimately tied to bonding models, representing the number of atoms chemically bonded to the central atom, which requires an understanding of the nature and strength of the interatomic interactions [15] [16]. The International Union of Crystallography (IUCr) explicitly acknowledges this duality, stating that the coordination number of an atom in a crystalline solid "depends on the chemical bonding model used" [15] [16]. This guide provides a comparative analysis of these two paradigms, examining their determination methods, underlying assumptions, and applications, with a focus on evaluating the accuracy of coordination number determination methods within modern research contexts.
Comparative Analysis: Geometric vs. Chemical Coordination Number
The following table summarizes the fundamental distinctions between geometric and chemical coordination numbers, highlighting their defining characteristics and methodological approaches.
Table 1: Core Distinctions Between Geometric and Chemical Coordination Numbers
| Feature | Geometric Coordination Number | Chemical Coordination Number |
|---|---|---|
| Fundamental Basis | Purely geometrical and topographical arrangement of atoms in space [15] | Chemical bonding model applied to the atomic structure [15] [16] |
| Primary Determination Method | Analysis of interatomic distances (e.g., Brunner-Schwarzenbach method, Voronoi-Dirichlet partitioning) [15] [16] | Analysis based on bonding interactions, often involving electron density analysis (e.g., QTAIM) and chemical intuition [15] |
| Dependence on Bonding Model | Independent; does not require a predefined bonding model [15] | Dependent; the value is defined by the chosen bonding model [16] |
| Coordination Reciprocity | Often not pairwise symmetric (e.g., atom A may be a neighbor of B, but not vice versa) [15] | Inherently pairwise symmetric due to the nature of a chemical bond [15] |
| Output Nature | Often an integer, but can be a weighted effective CN (ECoN) [16] | An integer count of chemical bonds |
| Example of Discrepancy | In the α-iron (BCC) structure, the geometric CN can be considered 8 (nearest neighbors) or 14 (including next-nearest) [16]. | From a chemical bonding perspective, the CN may be considered 8, focusing on the strongest interactions [16]. |
Experimental Protocols for Coordination Number Determination
Accurate determination of coordination numbers relies on specific experimental and computational protocols. The methodologies differ significantly depending on whether the goal is to extract geometric or chemical structural information.
Protocol for Geometric Coordination Number Analysis
The geometric coordination number is most commonly derived from crystallographic data obtained via X-ray, neutron, or electron diffraction experiments [16].
Step 1: Data Collection and Structure Refinement
- Procedure: A crystal of the material is analyzed via diffraction techniques. The resulting diffraction pattern is used to solve and refine the crystal structure, yielding a set of atomic coordinates, lattice parameters, and space group symmetry [15].
- Key Consideration: High-resolution data is crucial for accurate interatomic distance calculations.
Step 2: Interatomic Distance Calculation and Gap Analysis
- Procedure: A list of all interatomic distances from a central atom is generated and sorted in increasing order. The established Brunner-Schwarzenbach (BS) method is then applied to identify the first large gap in this distance sequence [15] [16].
- Mathematical Application: The gap signifies the boundary between the first coordination sphere and subsequent neighbors. The number of atoms before this gap is the geometric CN.
- Challenge: This method can produce non-reciprocal coordination (e.g., atom B is in A's coordination sphere, but A is not in B's) and may show multiple gaps of similar size, requiring a cut-off parameter [15].
Step 3: Voronoi-Dirichlet Partitioning (VDP) - An Alternative Geometric Method
- Procedure: Space is partitioned into polyhedral domains around each atom, comprising all points closer to that atom than to any other. Atoms whose Voronoi polyhedra share a face are considered neighbors [15].
- Result: The number of faces of the Voronoi polyhedron gives the maximum possible geometric CN. This method provides a unique, topology-based value but often overcounts compared to chemically expected coordination [15].
Protocol for Chemical Coordination Number Analysis
Determining the chemical coordination number moves beyond metrics to identify chemically meaningful bonds, which is particularly crucial in complex or disordered solids.
Step 1: Theoretical Electron Density Calculation
- Procedure: Employ quantum chemical calculations (e.g., Density Functional Theory) to obtain the theoretical electron-density distribution for the crystallographically determined structure. According to the Hohenberg-Kohn theorems, this density defines all ground-state properties [15].
- Objective: To move beyond purely geometric analysis to a quantum-mechanical basis for identifying atomic interactions.
Step 2: Topological Analysis via QTAIM
- Procedure: The Quantum Theory of Atoms in Molecules (QTAIM) is applied to the calculated electron density. In this framework, atomic basins are defined by zero-flux surfaces in the electron density gradient field. A bond path between two atoms indicates a chemical interaction [15].
- Innovation: Recent research utilizes triangulated surface data sets of QTAIM interatomic surfaces to calculate the solid angles subtended at each nuclear position. This allows for the calculation of a topological effective coordination number (tCN) that naturally includes the effect of different atomic sizes and satisfies coordination reciprocity [15].
Step 3: Coordination-Consistent Scenario Ranking
- Procedure: The tCN approach incorporates weighting functions to rank different possible coordination scenarios. This is vital in complex structures like intermetallic phases (e.g., TiNiSi type), where multiple coordination environments may be similarly plausible [15].
- Output: Instead of a single integer, the analysis may yield a list of different coordination scenarios with their relative weights and associated effective coordination numbers, providing a more nuanced chemical picture [15].
Visualization of Methodologies and Relationships
The following diagrams illustrate the core concepts and workflows involved in differentiating and determining coordination numbers.
Conceptual Relationship and Determination Pathways
Experimental Workflow for Modern Coordination Number Analysis
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagents and Computational Tools for Coordination Number Research
| Item | Function in Research |
|---|---|
| Single Crystals | High-quality, single-crystal samples are essential for obtaining high-resolution diffraction data required for accurate geometric and electron density analysis [16]. |
| X-ray Diffractometer | The primary instrument for experimental determination of crystal structures. Provides the atomic coordinate data that is the starting point for all coordination number analysis [16]. |
| Neutron Source | Provides neutron beams for neutron diffraction, which is particularly valuable for locating light atoms (e.g., H, Li) and distinguishing between elements with similar atomic numbers [16]. |
| Quantum Chemistry Software | Software packages (e.g., for DFT calculations) are used to compute the electron density distribution from the crystal structure, forming the basis for topological CN analysis [15]. |
| Topological Analysis Code | Specialized software (e.g., for QTAIM analysis) is required to perform topological analysis on the electron density to identify bond paths and calculate properties like the tCN [15]. |
The distinction between geometric and chemical coordination numbers is not merely semantic but represents a fundamental divergence in approach between purely descriptive crystallography and interpretive chemical bonding analysis. The geometric coordination number offers a reproducible, if sometimes chemically naive, metric that is invaluable for structural classification and database creation. In contrast, the chemical coordination number seeks to reflect the physical reality of bonding, a goal increasingly within reach through modern electron-density-based approaches like the topological coordination number (tCN). The emerging paradigm, especially for complex structures, is to move beyond a single integer and embrace a weighted, multi-scenario description of atomic environment [15]. This nuanced understanding is critical for researchers and drug development professionals who rely on accurate structural descriptors to rationalize material properties, reactivity, and biological activity, ultimately feeding into the development of more predictive AI models for structure-property relationships [15].
The precise characterization of atomic coordination environments is a cornerstone of modern materials science and biochemistry, directly impacting the development of advanced catalysts, functional materials, and therapeutic agents. Coordination numbers (CN) and the spatial arrangement of neighboring atoms dictate fundamental properties such as catalytic activity, mechanical strength, and biological function. However, accurately determining CNs remains challenging across complex systems including intermetallic compounds, single-atom catalysts (SACs), and biomolecules, where dynamic behavior, structural complexity, and limitations of analytical techniques introduce significant uncertainties. This review systematically compares experimental and computational methods for CN determination across these diverse systems, evaluating their accuracy, limitations, and applicability through direct comparison of published data and methodologies. By framing this analysis within the broader thesis of coordination number determination accuracy, we provide researchers with a critical framework for selecting appropriate characterization strategies based on their specific system requirements and accuracy constraints.
Fundamental Concepts and Definitions
Coordination Number in Context
The concept of coordination number originated from Werner's early work in coordination chemistry as a descriptor for the number of atoms directly bonded to a central atom [4]. In crystalline solids, CN represents a condensation of complete geometrical information about spatial atomic arrangements into a single numerical value specifying how many atoms inhabit the coordination environment of a central atom. While seemingly straightforward, the term lacks a universal mathematical definition, leading to methodological dependencies in its determination [4]. The International Union of Crystallography acknowledges that CN "depends on the chemical bonding model used," highlighting the tension between purely geometric and chemically meaningful definitions [4].
Methodological Approaches to CN Determination
Table 1: Comparison of Coordination Number Determination Methods
| Method | Fundamental Principle | Accuracy Limitations | Optimal Application Scope |
|---|---|---|---|
| Topological Effective CN (tCN) | Analysis of electron density distributions via QTAIM; calculates solid angles subtended at nuclear positions by diatomic contact surfaces | Natural inclusion of atomic size effects; requires high-quality electron density data | Complex intermetallic phases; systems with significant atomic size disparities |
| Voronoi-Dirichlet Partitioning (VDP) | Geometric partitioning of space into regions closer to each atom than any other | Overcounts neighbors (maximal CN); sensitive to metric data alone; violates coordination reciprocity | Preliminary analysis of high-symmetry structures |
| Brunner-Schwarzenbach (BS) | Identifies first large gap in interatomic distance sequence | Cut-off parameter dependence; often non-reciprocal (A in B's sphere but not vice versa) | Binary and simple ternary compounds with clear coordination gaps |
| X-ray Absorption Spectroscopy (XAS) | Measures fine structure of absorption edges to probe local coordination environment | Limited to ~0.02 nm distance resolution; requires reference compounds | SACs; dilute systems; in situ/operando studies |
| Nuclear Magnetic Resonance (NMR) | Probes local magnetic field environment and dynamics via relaxation profiles | Limited to NMR-active nuclei; interpretation complexity in paramagnetic systems | Biomolecular systems; dynamics over wide timescales |
The topological coordination number (tCN) approach represents a significant advancement by leveraging electron density distributions from the quantum theory of atoms in molecules (QTAIM) [4]. This method calculates solid angles subtended at nuclear positions by diatomic contact surfaces, effectively generalizing the VDP approach while naturally incorporating atomic size effects. Even in highly symmetrical elemental structures, differences between VDP and tCN results emerge due to atomic electron-density decay utilizing available degrees of freedom in the crystal structure [4]. For complex structures like TiNiSi-type compounds, tCN enables numerical ranking between different sub-coordination scenarios of similar importance, providing a more precise characterization through listing different scenarios with relative weights and associated effective coordination numbers [4].
Intermetallic Compounds
Structural Complexity and CN Challenges
Intermetallic phases exhibit beneficial combinations of high strength, low density, and corrosion resistance, making them ideal for high-temperature applications and severe environments [17]. The accurate determination of coordination environments in these systems is complicated by often complex structures, mixed bonding character, and the presence of multiple constituent elements with different atomic sizes and electronegativities.
In high-entropy intermetallics (HEIs), additional complexity arises from the presence of five or more elements in ordered structures. For example, in L1₀-ordered PtCoNiFeCu HEI catalysts, the distribution of constituent elements creates local atomic segregation and cluster formation, leading to sub-angstrom strain that significantly influences catalytic performance [18]. This strain results from displacement of transition metal atoms in the TM layers due to local clustering/segregation driven by the stabilizing effect of Pt layers [18].
Experimental Approaches and Key Findings
Table 2: Coordination Environment Analysis in Selected Intermetallic Systems
| Material System | Primary Characterization Methods | Key Findings on Coordination Environment | Performance Correlation |
|---|---|---|---|
| L1₀-PtCoNiFeCu HEI [18] | In situ XRD, XAFS, Rietveld refinement, atomic-resolution STEM | Sub-angstrom strain in TM layers; N-doping induces tensile strain (3.680±0.003Å vs 3.675±0.003Å) | Mass activity: 2.19 A mgₚₜ⁻¹ (MOR); Current density: 1388 mA cm⁻² at 0.7V after 90k cycles |
| Pt₃Co IMC [19] | Coordination-in-pipe engineering using SBA-15 templates, AC-HAADF-STEM, DFT | Particle size control (3-9 nm) via template diameter; coordination number manipulation via N-source | Mass activity: 2.19 A mgₚₜ⁻¹ for MOR (highest with 1,10-phenanthroline N source) |
| TiNiSi-type compounds [4] | Topological CN (tCN) via QTAIM electron density analysis | Multiple weighted sub-coordination scenarios; superior to VDP in complex structures | Input for AI applications predicting structure-property relationships |
Advanced synthesis methods enable precise control over coordination environments in intermetallics. The coordination-in-pipe engineering approach using SBA-15 templates allows simultaneous regulation of particle size (3-9 nm) and coordination environments in Pt-based intermetallic compounds (Pt-IMCs) [19]. This method confines metal precursors and nitrogen sources within the pipes of SBA-15 before pyrolysis, effectively hindering nanoparticle growth radially while nitrogen-doped carbon restricts longitudinal growth [19]. The coordination numbers between metal and nitrogen can be regulated using different N sources, significantly impacting catalytic performance [19].
The sub-angstrom strain in HEI catalysts introduces anisotropic strain that distinguishes them from binary intermetallic compounds [18]. This strain, combined with the pinning effect of metal-N bonds and the high-entropy effect, contributes to exceptional stability, providing current density of 1388 mA cm⁻² at 0.7 V after 90,000 cycles under heavy-duty vehicle conditions [18]. Nitrogen doping further amplifies these effects through interstitial doping, creating tensile strain evidenced by larger lattice parameters in N-HEI/KB (3.680 ± 0.003 Å) compared to HEI/KB (3.675 ± 0.003 Å) [18].
Single-Atom Catalysts (SACs)
Coordination Environment Significance
In single-atom catalysts, where isolated metal atoms are anchored on support materials, the coordination environment fundamentally determines catalytic performance. Unlike nanoparticles or intermetallic compounds, SACs lack metal-metal bonds, making the support-coordinate bonds exclusively responsible for stabilizing metal centers and modulating electronic properties. The coordination number in SACs typically refers to the number of heteroatoms (N, O, S, P) from the support directly bonded to the metal center, creating well-defined active sites with nearly 100% atom utilization.
Engineering Coordination Environments in SACs
Table 3: Coordination Engineering in Single-Atom Catalysts
| Catalyst System | Coordination Environment | Synthesis Strategy | Catalytic Performance |
|---|---|---|---|
| Pt-IMCs on N-doped C [19] | Pt-Nₓ coordination; tunable via N-source | Coordination-in-pipe engineering with SBA-15 templates | MOR mass activity: 2.19 A mgₚₜ⁻¹; enhanced due to high chemical states of Pt/Co |
| Cu-N-C SACs [20] | Cu-Nₓ sites in N-doped carbon | Pyrolysis of Cu precursors with N/C sources | Nitrate to ammonia conversion; performance depends on Cu speciation |
| M-N-C (M=Fe, Co, Ni) [20] | M-N₄ moieties in carbon matrix | Wet-impregnation or templating methods | ORR activity; stability challenges due to metal leaching |
Coordination engineering in SACs focuses on manipulating the number, identity, and spatial arrangement of atoms directly bonded to the metal center. The coordination-in-pipe engineering strategy demonstrates how both particle size and coordination environments can be simultaneously controlled in Pt-based catalysts [19]. By selecting appropriate nitrogen sources (e.g., 1,10-phenanthroline), the coordination number of interface metal atoms can be adjusted at the angstrom scale, directly influencing catalytic performance [19].
The chemical states of surface atoms, affected by nitrogen coordination number, facilitate electron accumulation on active sites, reduce activation energy of rate-determining steps, and enhance catalytic performance [19]. For Pt₃Co IMCs using 1,10-phenanthroline as the nitrogen source, the high chemical states of surface Pt and Co contribute to exceptional methanol oxidation reaction performance [19].
Biomolecular Systems
Coordination Complexity in Biological Contexts
Biomolecular coordination environments exhibit dynamic, often transient characteristics that complicate precise CN determination. In metalloproteins and supramolecular assemblies, metal centers frequently display coordination numbers that fluctuate in response to environmental conditions, substrate binding, and allosteric effects. The Cu/Zn/histidine supramolecular assemblies inspired by natural Cu-Zn superoxide dismutase (SOD) exemplify how coordination environment optimization can dramatically enhance biological function [21].
Experimentation and Analysis in Biomolecular Systems
Table 4: Biomolecular Coordination Environment Studies
| System | Experimental Methods | Coordination Environment Details | Functional Outcomes |
|---|---|---|---|
| Cu/Zn/His Supramolecular Assemblies [21] | Gibbs free energy calculations, SOD activity assays | Optimized Cu²⁺ site: 1 amino, 1 carboxyl, 2 imidazolyls (vs 4 imidazolyls in natural SOD) | SOD activity: 37,900 U/mg (5.4× natural SOD); promotes M1→M2 macrophage polarization |
| Biomolecular Dynamics [22] | Field-cycling NMR relaxometry, sample shuttling | Probing dynamics from ns-ms timescales; dipole-dipole, quadrupolar, CSA interactions | Atomic-resolution mobility mapping in near-physiological conditions |
In the CuZnHis system, researchers systematically compared different coordination modes for Cu²⁺ sites [21]. Calculations revealed that replacing two imidazolyl coordinations with one amino and one carboxyl group (Structure D) created an optimized coordination environment with lower overall energy and reduced energy for coordination bond breaking/reformation during catalysis [21]. This optimized coordination environment yielded dramatically enhanced SOD activity of 37,900 U/mg, at least 5.4 times higher than natural Cu-Zn-SOD [21].
NMR relaxometry provides unique insights into biomolecular coordination dynamics through field-dependent relaxation profiles [22]. This approach probes dynamic processes over wide timescales by measuring nuclear relaxation rates across magnetic fields from ~200 μT to over 100 MHz [22]. The technique is particularly valuable for characterizing metal coordination environments in paramagnetic biomolecular systems, where electron-nuclear interactions dominate relaxation mechanisms [22].
Comparative Analysis of Methodologies
Cross-System Method Evaluation
The accuracy of coordination number determination varies significantly across methodological approaches and system types. Topological CN (tCN) analysis based on electron density distributions offers particular advantages for complex intermetallic phases, where it naturally incorporates atomic size effects and enables numerical ranking of competing sub-coordination scenarios [4]. This approach avoids the coordination reciprocity problems inherent in traditional geometric methods like Brunner-Schwarzenbach, where atom A may be counted in B's coordination sphere but not vice versa [4].
For SACs and supported catalysts, X-ray absorption spectroscopy provides element-specific coordination information but requires careful interpretation and reference compounds [19] [18]. The coordination-in-pipe engineering approach demonstrates how synthetic control can manipulate coordination environments while providing built-in validation through systematic variation of template sizes and nitrogen sources [19].
In biomolecular systems, where coordination environments are dynamic and often transient, NMR relaxometry and thermodynamic analysis of coordination bond breaking/reformation offer insights into functional coordination numbers under near-physiological conditions [21] [22]. The CuZnHis system exemplifies how computational guidance (Gibbs free energy calculations of different coordination modes) can direct the design of optimized coordination environments with enhanced biological activity [21].
The Scientist's Toolkit
Table 5: Essential Research Reagents and Materials for Coordination Environment Studies
| Reagent/Material | Function in Coordination Studies | Representative Application |
|---|---|---|
| SBA-15 Templates | Mesoporous silica with tunable pore diameters (4-18 nm) for size-controlled synthesis | Confinement synthesis of Pt-IMCs with controlled particle size [19] |
| Nitrogen Sources (1,10-phenanthroline, etc.) | Coordination ligands that determine metal-N coordination number and chemical states | Adjusting coordination environments in Pt-IMCs for enhanced MOR activity [19] |
| Field-Cycling NMR Relaxometers | Instruments measuring nuclear relaxation rates from 0.01-100+ MHz Larmor frequency | Probing biomolecular dynamics over wide timescales [22] |
| Metal Precursors (Pt, Co, Ni, Fe, Cu salts) | Source of metal centers for intermetallic and SAC synthesis | Preparation of high-entropy intermetallic PtCoNiFeCu catalysts [18] |
| Histidine and Derivatives | Multidentate ligands for biomimetic coordination environments | Constructing optimized Cu catalytic sites in SOD-mimetic assemblies [21] |
This comparative analysis reveals that accurate coordination number determination requires method selection tailored to specific system characteristics and research objectives. For intermetallic compounds with complex structures, topological approaches leveraging electron density distributions provide the most chemically meaningful CNs. In single-atom catalysts, coordination engineering combined with spectroscopic validation enables precise manipulation of active sites. For biomolecular systems, dynamic analysis through techniques like NMR relaxometry captures functionally relevant coordination environments. Across all systems, the integration of multiple complementary methods and computational guidance offers the most robust approach to characterizing coordination environments and correlating them with functional properties. As coordination environment design continues to gain importance in developing advanced materials and therapeutics, methodological advances in accurate CN determination will remain crucial for establishing reliable structure-property relationships.
Modern Analytical Methods for Coordination Environment Characterization
Voronoi-Dirichlet partitioning is a fundamental geometric construction for analyzing atomic environments in crystallography and materials science. The method partitions space into convex polyhedra (Voronoi cells) around generating points (atomic nuclei), where each location within a cell is closer to its generating point than to any other. Mathematically, for a set of generators (P = {p1, p2, \ldots, pn}) in space (Z), the standard Voronoi cell is defined as (V(pi) = \bigcap{j \ne i} {p \in Z | d(p,pi) < d(p,p_j) }), where (d) is the Euclidean distance [23]. In the context of coordination number determination, these cells provide an intuitive geometric framework for identifying neighboring atoms and quantifying local atomic environments by analyzing the faces, edges, and vertices of the resulting polyhedra.
The method's significance extends across multiple scientific domains. In quantum crystallography, Voronoi-based analysis helps refine crystal structures beyond the independent atom model, enabling more accurate electron density mapping and chemical bonding analysis [24]. For materials informatics, it facilitates high-throughput screening of ion transport pathways in solid electrolytes and electrode materials by characterizing void space geometry [23]. Recently, Voronoi tessellations have been integrated into deep learning models for catalyst discovery, where they provide structurally constrained graph representations that improve property prediction accuracy [25].
Comparative Analysis of Voronoi Method Variants
Fundamental Methodologies and Their Applications
The standard Voronoi decomposition, while mathematically elegant, proves inadequate for many crystallographic applications where atoms have significantly different radii. This limitation has spurred the development of several variants, each designed to address specific challenges in structural analysis.
Table 1: Key Variants of Voronoi-Dirichlet Partitioning in Structural Science
| Method Variant | Mathematical Foundation | Primary Applications | Key Advantages | |
|---|---|---|---|---|
| Standard Voronoi | (V(pi) = \bigcap{j \ne i} {p \in Z | d(p,pi) < d(p,pj) }) [23] | Initial structure analysis, monodisperse systems | Computational simplicity, intuitive geometric interpretation |
| Radical Voronoi | (V(pi) = \bigcap{j \ne i} {p \in Z | d(p,pi)^2 - ri^2 < d(p,pj)^2 - rj^2 }) [23] | Crystals with atoms of unequal radii, ionic transport analysis | Accounts for atomic size differences, maintains convex polyhedra |
| Voronoi S | (V(pi) = \bigcap{j \ne i} {p \in Z | d(p,pi) - ri < d(p,pj) - rj }) [23] | Theoretical modeling of polydisperse systems | Most accurate representation of size-asymmetric systems |
| Residual-Weighted CVT | Adaptive sampling based on Voronoi tessellation with residual constraints [26] | Physics-Informed Neural Networks (PINNs) for PDE solutions | Improves prediction accuracy and stability in computational physics |
The radical Voronoi decomposition (also called Voronoi S-tessellation) represents a crucial advancement for practical crystallographic applications. It effectively compromises between the computational simplicity of the standard approach and the physical accuracy of the Voronoi S method, which produces curved boundaries that are computationally challenging to handle [23]. The radical method's ability to generate convex polyhedra while accounting for atomic radii makes it particularly suitable for analyzing structures with mixed elements, such as intermetallic compounds and ionic crystals.
Quantitative Performance Comparison
Evaluating the performance of these methods reveals significant differences in their practical utility for coordination environment analysis. The CAVD Python package, specifically designed for crystal structure analysis, implements radical Voronoi decomposition with environment-aware ionic radii to address coordination-dependent size variations [23].
Table 2: Performance Comparison of Voronoi-Based Methods for Structural Analysis
| Method Variant | Mobile Ion Site Recovery Rate | Computational Efficiency | Stability Across PDE Types |
|---|---|---|---|
| Standard Voronoi | Limited (excludes face-centered sites) [23] | High | Limited for nonlinear equations [26] |
| Radical Voronoi (CAVD) | 99% (6,955 ionic compounds) [23] | Moderate | Not specifically tested |
| Residual-Constrained V.T. (RVT) | Not applicable | Comparable to other sampling methods | Superior across various PDE problems [26] |
| Residual-Weighted CVT (RCVT) | Not applicable | Comparable to other sampling methods | Superior across various PDE problems and initialization conditions [26] |
The exceptional 99% recovery rate for mobile ion sites achieved by the CAVD tool demonstrates the critical importance of method selection. This performance advantage stems directly from its incorporation of Voronoi polyhedra faces as potential sites for mobile ions, whereas standard approaches typically consider only vertices and edges [23]. This comprehensive mapping of the void space enables more accurate prediction of ionic transport pathways in solid electrolytes and electrode materials.
Experimental Protocols and Methodologies
Implementation Workflow for Geometric Analysis
The standard experimental protocol for Voronoi-Dirichlet analysis in coordination number determination follows a systematic workflow implemented in tools like CAVD and ToposPro:
Figure 1: Experimental workflow for Voronoi-Dirichlet analysis in crystal structures
The process begins with crystal structure input followed by symmetry analysis using libraries like Spglib [23]. The critical atomic radius assignment step combines the rigorous coordination number definition by O'Keeffe with Shannon's effective ionic radii to account for coordination environment effects [23]. The core radical Voronoi decomposition is then performed, typically implemented via modified versions of the Voro++ library [23]. Finally, the resulting Voronoi network is analyzed to calculate structural descriptors and identify transport pathways.
Advanced Computational Frameworks
In machine learning applications, particularly for catalyst discovery, Voronoi tessellations enrich graph neural network representations. The experimental protocol involves:
- Graph Construction: Crystal structures are represented as graphs with atoms as nodes
- Voronoi Enhancement: Voronoi tessellation calculates solid angles and contact types as edge features, and Voronoi volumes as node characteristics [25]
- Property Prediction: The enhanced graph representation improves prediction of catalytic properties and formation energies [25]
This approach has demonstrated significant performance improvements, reducing the mean absolute error to 6 meV/atom on intermetallics datasets, well below the physically significant 20 meV/atom threshold [25].
Limitations and Methodological Constraints
Fundamental Geometric Limitations
Despite its mathematical elegance, Voronoi-Dirichlet partitioning exhibits several inherent limitations that affect its accuracy for coordination number determination:
Atomic Size Disparity: The standard Voronoi decomposition becomes increasingly inaccurate for structures containing atoms with significantly different radii [23]. This poses particular challenges for organometallic compounds and minerals containing heavy and light elements.
Dynamic Behavior Neglect: Traditional Voronoi analysis provides a static geometric snapshot, unable to capture thermal motion and dynamic disorder effects crucial for understanding ionic transport mechanisms [24].
Face-Centered Site Oversight: Standard Voronoi network approaches that consider only vertices and edges fail to identify mobile ion positions located on Voronoi polyhedra faces [23], potentially missing critical migration pathways.
Experimental Data Dependence: Quantum crystallographic applications using Voronoi-based methods remain heavily dependent on high-quality experimental data, as evidenced by dedicated efforts to obtain accurate temperature-dependent structure factors for reliable electron density analysis [24].
Implementation Challenges
Practical implementation of Voronoi methods faces several computational and methodological hurdles:
Radius Assignment Ambiguity: The accuracy of radical Voronoi methods depends critically on appropriate atomic radius selection, complicated by the dependence of ionic radii on coordination environments [23].
Boundary Approximation: The radical Voronoi approach, while more computationally tractable than the Voronoi S method, only approximates the true curved boundaries between atoms of different sizes [23].
High-Dimensional Extension: Current Voronoi-based sampling methods for physics-informed neural networks have proven effective for two-dimensional problems, but extension to three-dimensional cases requires further development [26].
Essential Research Tools and Reagents
Computational Infrastructure
Table 3: Essential Research Reagent Solutions for Voronoi-Dirichlet Analysis
| Tool/Resource | Function | Application Context |
|---|---|---|
| CAVD Python Package | Geometric analysis of void space in crystals | High-throughput screening of ionic transport materials [23] |
| Voro++ Library | Periodic radical Voronoi decomposition | Core computational engine for space partitioning [23] |
| Spglib | Symmetry analysis of crystal structures | Preprocessing for Voronoi tessellation [23] |
| Shannon Ionic Radii | Effective ionic radii database | Atomic size parameterization for radical Voronoi [23] |
| Open Catalyst Project Dataset | Benchmark dataset for catalytic properties | Validation of Voronoi-enhanced machine learning models [25] |
The CAVD Python package exemplifies the modern implementation of Voronoi-based structural analysis, specifically designed for high-throughput ion-transport analysis and freely available to the research community [23]. It builds upon the computational geometry capabilities of the Voro++ library while adding chemistry-aware functionality through proper atomic radius assignment and consideration of Voronoi faces in network construction.
Specialized Methodological Components
For quantum crystallographic applications, specialized tools have been developed to address specific refinement challenges:
- Hirshfeld Atom Refinement (HAR): Integrates quantum chemical electron densities with X-ray diffraction data, moving beyond the independent atom model [24]
- NoSpherA2: Implements effective core potentials with ZORA to accelerate refinement of heavy-element structures [24]
- expHAR: Alternative electron density partition scheme that improves hydrogen position accuracy [24]
- ReCrystal: Employs periodic DFT-derived multipoles for tailored refinement without external libraries [24]
These tools represent the cutting edge of crystallographic refinement, addressing fundamental limitations of traditional independent atom model approaches that still dominate routine structure determination.
Voronoi-Dirichlet partitioning remains an indispensable geometric approach for coordination number determination and structural analysis across crystallography, materials science, and computational chemistry. The method's fundamental strength lies in its intuitive geometric representation of atomic environments and void space topology. The development of radical Voronoi variants has significantly addressed the critical limitation of atomic size disparity, enabling accurate analysis of complex multi-element systems as demonstrated by the 99% site recovery rate achieved by the CAVD tool.
Nevertheless, important limitations persist, particularly regarding the treatment of dynamic processes, electron density deformations in chemical bonds, and computational challenges in high-dimensional applications. The ongoing integration of Voronoi-based descriptors into machine learning frameworks represents a promising direction, combining geometric intuition with data-driven pattern recognition. As quantum crystallography continues to advance beyond the independent atom model, Voronoi-inspired methodologies will likely play an increasingly important role in bridging experimental measurement and quantum mechanical theory, ultimately enabling more accurate structure-property relationships for materials design and drug development.
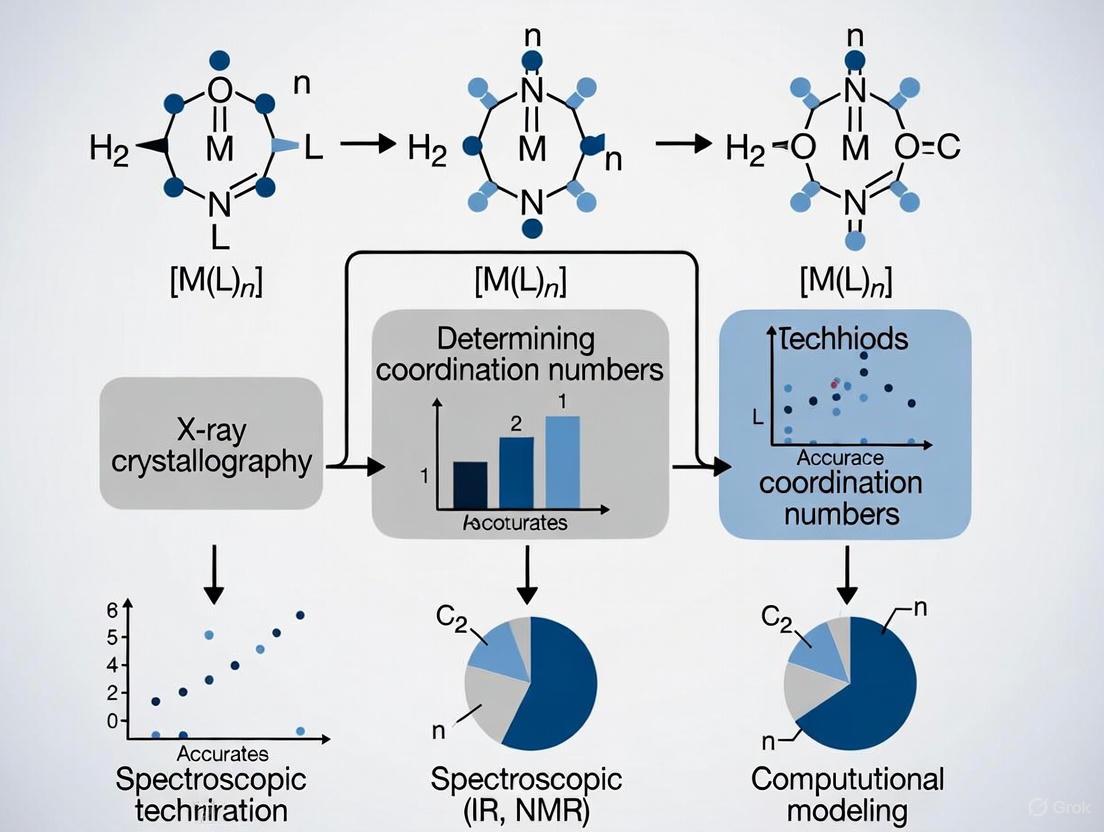
Topological Coordination Numbers from Electron Density Distributions
In crystallography and materials science, the coordination number (CN) is a fundamental descriptor that condenses the complex spatial arrangement of atoms surrounding a central atom into a single number. Traditionally, this parameter has been determined through geometric approaches analyzing interatomic distances or Voronoi partitioning. However, these methods possess significant limitations, including coordination reciprocity violations where atom A may be counted in the coordination sphere of B, but not vice versa, despite the same interatomic distance. This inconsistency presents substantial challenges for physical chemistry applications where bonding interactions are inherently pairwise symmetric [15].
The emerging paradigm of quantum crystallography, which celebrates the centenary of quantum mechanics in 2025, bridges crystallographic experimentation with quantum theoretical approaches [24]. Within this framework, a novel methodology has been developed: topological coordination numbers (tCNs) derived from electron density distributions. This approach leverages the quantum theory of atoms in molecules (QTAIM) to provide a more physically meaningful characterization of atomic coordination that obeys coordination reciprocity principles [15] [27]. This analysis is particularly valuable for understanding complex structures such as intermetallic phases and represents a significant advancement in digitizing structural information for artificial intelligence applications predicting structure-property relationships [15].
Methodological Comparison: Traditional vs. Topological Approaches
Established Coordination Number Determination Methods
Traditional CN evaluation methods rely primarily on geometric information derived from crystal structures:
- Distance-Based Methods: The Brunner-Schwarzenbach (BS) approach identifies the first large gap in the sequence of increasing interatomic distances. Used in major databases like Pearson's Crystal Data, this method suffers from subjectivity in gap identification and frequent reciprocity violations [15].
- Voronoi-Dirichlet Partitioning (VDP): This geometric construction defines spatial domains around each atom, with coordination determined by polyhedra sharing faces. While more systematic, VDP typically overcounts coordination (yielding higher CNs than chemically expected) and doesn't naturally account for atomic size differences [15].
- Effective Coordination Numbers (ECoN): This refinement applies weights to neighbors based on solid angles or other parameters, but often requires empirical corrections [15] [28].
Topological Coordination Number (tCN) Methodology
The tCN approach represents a paradigm shift by using the fundamental electron density distribution rather than purely geometric considerations:
- Foundation in Electron Density: According to the Hohenberg-Kohn theorems, all ground-state properties of a system, including structure, are determined by its electron density distribution [15].
- QTAIM Framework: The method employs triangulated surface data sets of QTAIM interatomic surfaces to calculate solid angles subtended at nuclear positions by each diatomic contact surface [15] [29].
- Coordination Reciprocity: A fundamental advantage is built-in pairwise symmetry, ensuring that if atom A coordinates to B, then B also coordinates to A—a physical requirement often violated in traditional methods [15] [27].
- Atomic Size Effects: Unlike VDP, the tCN approach naturally incorporates different atomic sizes through their distinct electron density decay profiles [15].
- Multiple Scenarios with Weights: Rather than forcing a single CN value, tCN analysis can identify and rank multiple chemically plausible sub-coordination scenarios with associated weights, providing a more nuanced description, especially in complex structures [15].
Table 1: Comparison of Coordination Number Determination Methods
| Method | Fundamental Basis | Reciprocity Guaranteed? | Accounts for Atomic Size? | Primary Limitations |
|---|---|---|---|---|
| Brunner-Schwarzenbach | Interatomic distances | No | No | Multiple gaps of similar size; cutoff parameter dependent |
| Voronoi-Dirichlet | Geometric space partitioning | Yes (by face sharing) | No (without corrections) | Overcounts coordination; purely geometric |
| Effective Coordination (ECoN) | Weighted solid angles/overlaps | Not necessarily | With parameterization | Requires empirical corrections |
| Topological CN (tCN) | Electron density distribution (QTAIM) | Yes | Yes, naturally | Computationally more demanding |
Experimental Protocols and Workflows
Theoretical Foundation and Computational Framework
The tCN methodology builds upon the quantum theory of atoms in molecules (QTAIM) developed by Bader [15] [29]. The core principle involves analyzing the topology of the electron density distribution ρ(r), particularly its gradient vector field ∇ρ(r) and Laplacian ∇²ρ(r). Atomic basins are defined by zero-flux surfaces satisfying ∇ρ(r)·n(r) = 0, where n(r) is the unit vector normal to the surface. Bond critical points (BCPs) occur where ∇ρ(r) = 0, and atomic interaction lines connecting nuclei through BCPs define the molecular graph representing chemical bonding [29].
For tCN determination, the key innovation involves calculating solid angles subtended by QTAIM interatomic surfaces at nuclear positions. These solid angles provide a quantitative measure of the "share" each neighbor has in the coordination sphere. The topological effective coordination numbers are then derived by summing these contributions according to specific weighting functions based on geometrical properties of square and semicircle areas [15].
Workflow for tCN Determination
The following diagram illustrates the comprehensive workflow for determining topological coordination numbers from initial structure to final coordination analysis:
Workflow for tCN Determination
Application to Material Classes
The tCN approach has been validated across diverse structural types:
- Elemental Structures: Face-centered cubic (fcc), body-centered cubic (bcc), hexagonal close-packed (hcp), and diamond-type structures [15].
- Binary Compounds: Rocksalt, CsCl, and zincblende structure types [15].
- Complex Intermetallics: TiNiSi-type structures and other complex phases where multiple coordination scenarios coexist [15].
- Dense Chalcogenides: AIVXVI and AV2XVI3 families (e.g., GeTe, PbSe, Bi2Te3) where coordination deviates from the classical 8-N rule [28].
For complex structures like TiNiSi-type compounds, the tCN advantage becomes particularly evident. Instead of selecting one possibly arbitrary coordination scenario, the analysis provides a weighted list of different chemically meaningful scenarios, offering a more comprehensive characterization [15].
Comparative Performance Analysis
Quantitative Comparison Across Structure Types
The tCN method demonstrates significant differences compared to traditional approaches, even in highly symmetric elemental structures, due to its incorporation of atomic electron-density decay characteristics:
Table 2: Performance Comparison of CN Methods Across Material Systems
| Material System | Traditional CN | Topological CN | Key Advantages of tCN |
|---|---|---|---|
| Face-Centered Cubic Elements | 12 (VDP) | Often <12 | Accounts for electron density anisotropy |
| Body-Centered Cubic Elements | 8 (VDP) or 6+8 (BS) | Intermediate values | Resolves coordination ambiguity naturally |
| Diamond-Type Structures | 4 (all methods) | 4 with reciprocity | Confirms conventional assignment |
| Rocksalt Structures | 6 (all methods) | 6 with reciprocity | Validates simple cases |
| TiNiSi-Type Compounds | Single value | Multiple weighted scenarios | Reveals complex coordination chemistry |
| Dense Chalcogenides (e.g., PbTe) | 6 (geometric) | Effective >6 with multicenter bonding | Identifies electron-deficient multicenter bonds [28] |
Resolution of Coordination Ambiguities
A particularly compelling advantage emerges in structurally complex intermetallic compounds:
- Multiple Scenarios: In TiNiSi-type structures, tCN analysis identifies several sub-coordination scenarios of similar importance, each with associated weights and effective coordination numbers [15].
- Beyond Binary Decisions: Rather than forcing a binary choice between competing coordination environments, tCN provides a quantitative ranking, acknowledging the complexity of chemical bonding in these systems [15].
- AI/ML Compatibility: This weighted, multi-scenario characterization provides richer, more nuanced input features for machine learning models predicting material properties [15] [30].
Implementing topological coordination number analysis requires specialized software tools and computational resources:
Table 3: Essential Research Tools for Topological Coordination Analysis
| Tool/Resource | Type | Primary Function | Application in tCN Analysis |
|---|---|---|---|
| CRITIC2 | Software Package | Topological analysis of electron density | Core QTAIM analysis; identification of interatomic surfaces [28] |
| Quantum Espresso | Software Package | Density functional theory calculations | Electron density distribution calculation [31] |
| ToposPro | Software Package | Topological analysis of crystal structures | Complementary topological analysis; 'color' topological types [32] |
| LOBSTER | Software Package | Local orbital basis suite for electronic structure | Orbital-based bonding analysis (alternative perspective) [28] |
| Experimental Charge Density | Methodology | Multipole refinement of diffraction data | Experimental electron density for tCN calculation [24] |
| Hirshfeld Atom Refinement (HAR) | Refinement Method | Quantum-crystallographic structure refinement | Improved accuracy for light atoms (hydrogen) [24] |
Implications for Materials Discovery and Design
The tCN methodology extends beyond academic interest to practical applications in materials design:
- Rational Materials Design: In dense chalcogenides, tCN analysis helps identify electron-deficient multicenter bonds (EDMBs), explaining anomalous sixfold coordination that defies the conventional 8-N rule. This understanding enables targeted design of phase-change materials and thermoelectrics [28].
- Topological Materials Development: For topological insulators and semimetals, accurate coordination characterization aids in understanding and predicting surface state behavior, crucial for next-generation electronic interconnects [31].
- Glass Science: Topological constraint theory combined with machine learning uses coordination numbers to predict glass properties (transition temperature, thermal expansion), accelerating discovery of functional glasses for electronic devices [30].
- High-Entropy Alloys: 'Color' topological type analysis helps model disordered structures by identifying topologically non-equivalent atomic nets, significantly reducing configurations needed for DFT modeling [32].
Topological coordination numbers derived from electron density distributions represent a significant advancement in accurately characterizing atomic environments in crystalline materials. By building upon the rigorous foundation of QTAIM and incorporating coordination reciprocity as a fundamental requirement, the tCN approach resolves longstanding limitations of traditional geometric methods.
The key advantages of this methodology include its physically grounded basis in electron density, natural accommodation of atomic size effects, consistent coordination reciprocity, and ability to characterize complex environments through weighted multiple scenarios. These features make tCN analysis particularly valuable for understanding contemporary material systems, including intermetallics, phase-change materials, topological conductors, and complex functional materials.
As quantum crystallography continues to mature, topological coordination number analysis stands as a powerful tool for bridging experimental crystallography with quantum mechanical principles, ultimately enabling more rational design of materials with tailored properties. The digitized, AI-compatible output of tCN characterization further positions this methodology as a key component in the evolving landscape of computational materials discovery and design.
X-ray Absorption Spectroscopy (XAS) is a powerful analytical technique that provides element-specific information about the local electronic structure and atomic environment around a selected element in a material. As a core technique at synchrotron radiation facilities, XAS has attracted diverse scientific communities from materials science to pharmaceutical research [33] [34]. The technique is based on measuring the absorption coefficient of a material as a function of the incident X-ray photon energy, particularly near and above an element's characteristic absorption edge [34]. This element-specificity makes XAS uniquely powerful for probing the local environment of a particular atomic species without interference from the surrounding matrix [34]. The technique is broadly divided into two main regions: X-ray Absorption Near-Edge Structure (XANES) and Extended X-ray Absorption Fine Structure (EXAFS), which provide complementary information about the absorbing atom's environment [35] [36].
The fundamental physical process underlying XAS involves the ejection of a core-level electron when the energy of an incident X-ray photon equals or exceeds the binding energy of that electron [34]. This photoelectric effect creates a core hole that is subsequently filled through electronic relaxation processes, and the measurement of these interactions provides rich information about the absorbing atom's chemical state and local coordination environment [34] [36]. Unlike X-ray diffraction methods that require long-range order in materials, XAS is uniquely capable of probing local structures in both crystalline and amorphous materials, including solids, liquids, and gases, without special sample preparation requirements [34] [36]. This versatility, combined with its sensitivity to oxidation states, coordination numbers, and bond distances, has established XAS as an indispensable tool for characterizing materials across numerous scientific disciplines [35] [36].
Technical Comparison of XAS, XANES, and EXAFS
Fundamental Principles and Physical Origins
XAS, XANES, and EXAFS originate from the same physical phenomenon but probe different aspects of the local atomic environment through distinct regions of the absorption spectrum. The entire XAS spectrum encompasses both the XANES region (extending from a few eV below to about 50 eV above the absorption edge) and the EXAFS region (extending from about 50 eV to as much as 1000 eV above the edge) [36]. The XANES region, sometimes referred to as Near-Edge X-ray Absorption Fine Structure (NEXAFS), is dominated by electronic transitions to unoccupied states and multiple scattering resonances [35] [36]. These features provide a "fingerprint" of the electronic structure and coordination geometry around the absorbing atom, reflecting the density of unoccupied states, oxidation state, and site symmetry [35] [37].
In contrast, the EXAFS region originates from the interference patterns created when the photoelectron wave emitted from the absorbing atom is scattered back by neighboring atoms [35] [36]. These constructive and destructive interference patterns manifest as oscillations in the absorption coefficient above the absorption edge. While XANES is sensitive to electronic structure and three-dimensional coordination geometry, EXAFS primarily provides quantitative information about interatomic distances, coordination numbers, and the chemical identities of neighboring atoms within approximately 5-6 Å of the absorbing atom [36]. The physics governing these spectral regions differs significantly: XANES involves multiple scattering events and lacks closed-form analytical expressions, making it more challenging to interpret quantitatively, whereas EXAFS can be described using scattering path expansions and is more analytically tractable [38] [37].
Comparative Analysis of Capabilities and Limitations
Table 1: Technical comparison of XAS spectral regions and their information content
| Parameter | XANES | EXAFS |
|---|---|---|
| Spectral Range | ±30-50 eV around absorption edge | 50-1000 eV above absorption edge |
| Primary Information | Oxidation state, coordination symmetry, electronic structure | Interatomic distances, coordination numbers, neighbor identity |
| Physical Origin | Electronic transitions to unoccupied states & multiple scattering | Single scattering of photoelectrons from neighboring atoms |
| Bond Distance Precision | Qualitative/semi-quantitative (~0.04-0.1 Å) | High precision (~0.01-0.02 Å) |
| Coordination Number Accuracy | ~20-30% | ~10-20% in favorable cases |
| Theoretical Treatment | Multiple scattering theory, DFT, real-space Green's functions | Single scattering theory, Fourier transform techniques |
| Sample Requirements | Can be applied to all phases (solid, liquid, gas) | Requires reasonable signal-to-noise for oscillation analysis |
| Key Limitations | Complex theoretical interpretation, sensitive to multiple parameters | Limited to ~5-6 Å range, challenging for heterogeneous bond lengths |
The complementary nature of XANES and EXAFS makes them powerful when used together for comprehensive local structure characterization. XANES serves as an excellent qualitative tool for fingerprinting chemical states and coordination environments, particularly through comparison with reference compounds [35]. The energy position of the absorption edge in XANES provides direct information about the oxidation state of the absorbing atom, with higher oxidation states typically shifting the edge to higher energies [35]. The pre-edge features in XANES spectra offer insights into electronic transitions and are particularly sensitive to coordination geometry and symmetry-breaking effects [35] [37].
EXAFS, on the other hand, delivers quantitative structural parameters with high precision for bond distances (typically 0.01-0.02 Å) and reasonable accuracy for coordination numbers (10-20% in favorable cases) [36]. The Fourier transform of the EXAFS oscillations converts the data from energy space to real space, producing a radial distribution function that reveals the distances and coordination numbers of neighboring atomic shells around the absorber [35]. However, EXAFS analysis becomes increasingly challenging when dealing with heterogeneous bond lengths or when the absorber is surrounded by light elements that are weak backscatterers [36]. Both techniques share the advantage of being applicable to a wide range of sample types, including crystalline solids, amorphous materials, liquids, and gases, without requiring long-range order [34] [36].
Experimental Methodologies and Protocols
Data Collection Techniques and Measurement Geometries
XAS experiments employ different measurement geometries optimized for specific sample types and concentration ranges. The three primary detection modes are transmission, fluorescence, and electron yield, each with distinct advantages and limitations [34]. Transmission mode, the most straightforward geometr,y involves measuring the intensity of the X-ray beam before (I₀) and after (Iₜ) it passes through the sample using ionization chambers [34] [36]. This method provides high-quality spectra with short acquisition times but requires homogeneous samples with optimal thickness and an absorber concentration typically exceeding 10% [34]. The optimal sample thickness for transmission measurements follows the relationship μd ≈ 1, where μ is the absorption coefficient and d is the sample thickness, to maximize signal-to-noise while maintaining reasonable transmitted intensity [36].
For dilute samples or those with low concentrations of the element of interest, fluorescence detection mode is preferred [34] [36]. This geometry measures the intensity of characteristic X-ray fluorescence (I_f) emitted when core holes created during the absorption process are filled by electrons from higher shells [34]. In typical fluorescence geometry, the incident X-ray beam and detector are positioned at 45° relative to the sample surface normal, minimizing elastic scattering background [34]. Fluorescence mode is particularly valuable for studying trace elements (down to hundreds of ppm) in complex matrices, such as biological samples or catalysts with low metal loadings [36]. However, fluorescence measurements can be affected by self-absorption effects, especially for concentrated samples or when the energy of the incident radiation is close to the absorption edge [34]. For such cases, specialized detection methods like partial fluorescence yield or total fluorescence yield can be employed, or data can be corrected during processing using software such as ATHENA [34].
Electron yield detection measures the current generated by electrons emitted during the relaxation processes following photoionization [36]. This method is particularly surface-sensitive (typically probing depths of ~5-50 nm, depending on the electron escape depth) and is often used for studying thin films or surface phenomena [36]. Each detection method offers complementary advantages, and the choice depends on factors such as sample concentration, homogeneity, physical state, and the specific information required.
Data Analysis Workflows and Computational Approaches
The analysis of XAS data follows systematic workflows that transform raw absorption measurements into meaningful structural parameters. For EXAFS analysis, the standard procedure involves several sequential steps: pre-edge background subtraction, post-edge background removal, normalization, conversion from energy to photoelectron wavevector (k-space), and Fourier transformation to real space (R-space) [35]. The resulting χ(k) oscillations and their Fourier transforms provide the basis for quantitative structural fitting using theoretical standards generated from programs such as FEFF, which calculates photoelectron scattering paths based on assumed structural models [35] [37].
XANES analysis employs both qualitative and quantitative approaches. Qualitative analysis includes fingerprint comparisons with reference compounds, examination of edge shifts for oxidation state determination, and analysis of pre-edge features for coordination geometry information [35]. Quantitative XANES analysis may involve linear combination fitting (LCF) to determine the relative proportions of known phases in a mixture, principal component analysis (PCA) to identify the number of significant components in a dataset, and multivariate curve resolution to extract spectral profiles of individual components [39] [35]. For both EXAFS and XANES, the development of machine learning approaches has revolutionized data analysis, enabling rapid identification of coordination environments and prediction of structural parameters directly from spectra [38] [40] [41].
Table 2: Comparison of traditional and machine learning approaches for XAS data analysis
| Analysis Aspect | Traditional Methods | Machine Learning Approaches |
|---|---|---|
| Coordination Environment Identification | Reference compound comparison, fingerprinting | Random forest models (>80% accuracy) [40], neural networks |
| Bond Distance Determination | EXAFS curve fitting with theoretical standards | Unified regression models (e.g., XAStruct) [38] |
| Oxidation State Analysis | Edge position measurement, linear combination analysis | Spectral feature extraction and pattern recognition |
| Data Processing Speed | Minutes to hours per spectrum | Near real-time (seconds) for trained models |
| Element Transferability | Limited, requires element-specific standards | Models spanning >70 elements (e.g., XAStruct) [38] |
| Software Tools | Athena, Artemis, FEFF, LARCH | XASDAML [41], PyFitIt, XANESNET, TRixs |
Diagram 1: XAS data analysis workflow showing parallel pathways for EXAFS and XANES analysis
Accuracy Assessment for Coordination Number Determination
Quantitative Performance Metrics Across Techniques
The accuracy of coordination number determination varies significantly between XANES and EXAFS and depends on numerous experimental and analytical factors. EXAFS typically provides coordination numbers with an accuracy of 10-20% under favorable conditions, though this can deteriorate to 20-30% or worse for complex systems with heterogeneous bond lengths, high disorder, or when light elements are involved [36]. The precision of EXAFS-derived bond distances is generally much better (0.01-0.02 Å) than that of coordination numbers because distances primarily affect the frequency of the EXAFS oscillations, while coordination numbers influence their amplitude, which is more susceptible to various experimental artifacts [36]. For XANES, coordination number estimation is generally more qualitative, often achieved through comparison with reference compounds of known structure or using advanced multivariate analysis methods [35].
Several factors limit the accuracy of coordination number determination in EXAFS. The presence of structural and thermal disorder (σ², the Debye-Waller factor) is convoluted with coordination number in the EXAFS amplitude, making it challenging to separate these contributions [35] [36]. Inelastic losses and many-body effects reduce the amplitude of EXAFS oscillations by approximately 10-20%, and if not properly accounted for, can lead to systematic underestimation of coordination numbers [37]. For first-shell coordination analysis, errors can be minimized by working at low temperatures to reduce thermal disorder, using appropriate amplitude reduction factors (S₀²), and analyzing multiple edges or multiple standards when available [35] [36].
Methodological Comparisons and Validation Studies
Recent advances in machine learning have demonstrated remarkable accuracy in coordination environment identification from XAS data. Random forest models trained on large computed XANES datasets (190,000 K-edge spectra) have achieved 85.4% accuracy in identifying main atomic coordination environments and 81.8% Jaccard score for all associated coordination environments across 33 cation elements in oxides [40]. These data-driven approaches significantly outperform traditional fingerprinting methods and maintain high accuracy when applied to experimental spectra [40]. The XAStruct framework represents another major advancement, providing a unified model for mean nearest-neighbor distance prediction that generalizes across more than 70 elements without element-specific tuning [38]. This machine learning approach addresses the long-standing inverse problem in XAS analysis by directly inferring local structural descriptors from spectral input.
Table 3: Accuracy comparison for coordination number determination methods
| Method | Coordination Number Accuracy | Bond Distance Precision | Elements Validated | Limitations |
|---|---|---|---|---|
| Traditional EXAFS | 10-20% (favorable cases) | 0.01-0.02 Å | Universal | Degrades with disorder, limited to ~5-6 Å range |
| XANES Fingerprinting | Qualitative (~20-30%) | 0.04-0.1 Å | Universal | Requires appropriate reference compounds |
| Linear Combination Fitting | 5-15% (with good references) | N/A | Universal | Dependent on reference quality and completeness |
| Random Forest ML [40] | >80% classification accuracy | N/A | 33 cation elements | Limited to trained coordination motifs |
| XAStruct Framework [38] | Unified MNND prediction | Regression model | >70 elements | Separate models per element for categorical data |
Validation studies comparing XAS-derived coordination numbers with known crystal structures consistently show that accuracy depends strongly on the data quality (signal-to-noise, k-range), the similarity between the unknown and reference compounds, and the care taken during data analysis [35] [36]. For XANES analysis, the pre-edge and main-peak regions have been identified as particularly important for coordination environment identification through drop-variable feature importance analysis [40]. The integration of machine learning with traditional EXAFS analysis has shown promise in overcoming some limitations of conventional approaches, particularly through their ability to identify complex patterns in high-dimensional spectral data that may be difficult to recognize through manual analysis [38] [41].
Advanced Applications in Pharmaceutical Sciences
Drug Development and Bioavailability Studies
XAS techniques have found important though still limited applications in pharmaceutical research, particularly in studying metal-containing drugs and their behavior in biological systems. The element-specificity of XAS enables researchers to probe the local environment of metal atoms in complex pharmaceutical formulations without interference from organic matrices [34]. This capability has been leveraged to study interactions between components in parenteral nutrition solutions, particularly zinc and amino acids, which can modify their bioavailability [42]. In these studies, EXAFS provided detailed information about coordination environments and binding modes that directly impact drug stability and delivery [42].
Another significant application involves the characterization of binary and ternary copper-amino acid complexes for developing efficient oral drugs against copper deficiencies in Menkes disease [42]. EXAFS and XANES analysis allowed researchers to characterize the solution structures of these complexes, providing insights critical for optimizing their therapeutic efficacy. Similarly, XAS has been used to characterize the solution form of arsenic-containing drugs for leukemia treatment, revealing how the local atomic structure around the arsenic center influences drug activity and toxicity profiles [42]. These applications demonstrate the unique capability of XAS to provide structural information about metal-based pharmaceuticals in relevant solution states, which is often difficult to obtain with other techniques [34] [42].
Drug-Biomolecule Interactions and Formulation Analysis
The high sensitivity of XAS to chemical state and local coordination environment makes it particularly valuable for studying drug-biomolecule interactions and differences in drug activity [34]. Modern XAS studies using synchrotron radiation enable real-time monitoring of structural changes in drugs, investigation of drug-DNA interactions, and characterization of metal coordination in proteins [34]. These capabilities are enhanced by the high penetration depth of X-ray radiation, which allows studies of samples in solid, liquid, or gaseous states without special preparation [34]. The independence from long-range order makes XAS suitable for analyzing both crystalline and amorphous pharmaceutical materials, a significant advantage when studying poorly crystalline active pharmaceutical ingredients (APIs) or disordered drug formulations [34].
Despite these promising applications, XAS remains underutilized in pharmaceutical research compared to fields like materials science and catalysis [34]. This underutilization stems partly from the technical challenges associated with synchrotron-based measurements and the expertise required for data interpretation. However, recent advances in table-top XAS instruments and user-friendly data analysis software are making the technique more accessible to pharmaceutical researchers [34] [41]. The growing recognition of metals' important roles in biological systems and pharmaceutical compounds suggests that XAS applications in drug development will expand significantly in the coming years, particularly for characterizing metallodrugs, tracing metal distributions in tissues, and optimizing metal-containing drug formulations [34] [42].
Essential Research Tools and Reagent Solutions
The XAS research landscape features several essential software tools that enable data processing, analysis, and interpretation. Demeter software suite (including Athena and Artemis) provides comprehensive capabilities for processing XAS data, performing background subtraction, normalization, and EXAFS fitting [39]. These programs implement standard data analysis procedures and are widely used in training workshops and by experienced researchers alike [39]. For theoretical calculations and spectral simulation, FEFF stands as a cornerstone code that performs real-space multiple-scattering calculations of XAS spectra using ab initio potentials [37]. FEFF enables quantitative interpretation of both EXAFS and XANES by calculating photoelectron scattering paths and has been extensively validated against experimental data [37].
Recent years have witnessed the development of machine learning frameworks that complement traditional XAS analysis software. XASDAML represents one such platform that integrates the entire data processing workflow, from spectral-structural descriptor generation to predictive modeling and performance validation [41]. This open-source framework, accessible through Jupyter Notebook interfaces, enables researchers to generate custom XAS datasets, build machine learning models, and obtain structural predictions from experimental data [41]. Similarly, the XAStruct framework provides a unified approach for both predicting XAS spectra from crystal structures and inferring local structural descriptors from spectral input, with demonstrated capability spanning over 70 elements across the periodic table [38]. These tools collectively enhance research efficiency and make advanced XAS analysis more accessible to non-specialists.
Experimental Reagents and Reference Materials
Table 4: Essential research reagents and materials for XAS experiments
| Material/Reagent | Function | Application Examples |
|---|---|---|
| Ionization Chambers | Measure X-ray intensity before/after sample | Standard detectors in transmission geometry |
| Fluorescence Detectors | Detect characteristic X-ray emission | Dilute samples, trace metal analysis |
| Reference Foils | Energy calibration, reference spectra | Metal foils (Cu, Fe, Zn) for edge calibration |
| Diluent Materials | Sample preparation for transmission mode | Boron nitride, cellulose, polyethylene |
| Sample Holders | Containment for various sample types | Liquid cells, pellet holders, cryostats |
| Beamline Components | Monochromate and focus X-ray beam | Si(111), Si(220) crystals; mirrors, slits |
Successful XAS experiments require appropriate reference materials for energy calibration and spectral interpretation. Metal foils (typically 5-10 μm thick) of pure elements corresponding to the absorption edge being studied are essential for energy calibration [36]. For quantitative analysis, well-characterized reference compounds with known structures similar to the unknown samples provide crucial benchmarks for both XANES fingerprinting and EXAFS analysis [35]. In pharmaceutical applications, this may include metal-organic complexes, metalloproteins, or model compounds that mimic expected coordination environments [34] [42]. For sample preparation, chemically inert diluents like boron nitride, cellulose, or polyethylene are used to achieve optimal sample thickness for transmission measurements, while specialized holders maintain sample integrity under various experimental conditions (cryogenic temperatures, controlled atmospheres, etc.) [36]. The continued development of these research tools and reference materials remains essential for advancing the application of XAS techniques across scientific disciplines, including the growing field of pharmaceutical research.
Continuous Symmetry Measures for Quantifying Geometric Deviations
This guide provides an objective comparison of methods for quantifying geometric deviations from ideal symmetry, with a specific focus on their application in evaluating coordination number determination accuracy. Continuous Symmetry Measures (CSMs) transform symmetry from a binary property into a quantifiable descriptor, enabling precise analysis of molecular structures and coordination environments crucial for materials science and drug development.
The core of Continuous Symmetry Measures involves calculating the minimal deviation required for a molecular structure to achieve a specified perfect symmetry. The fundamental measure is a numerical value between 0 and 100, where 0 represents perfect symmetry [43].
Several computational strategies have been developed to handle structures of varying size and complexity [43]:
Exact Algorithm: This method scans all mathematically possible permutations of atoms to find the nearest symmetric structure. It is computationally intensive and is generally feasible only for small-to-medium molecules. For larger systems, a variation that scans only "structure-preserving permutations" (those that maintain the original chemical connectivity) can be used to make the calculation tractable while retaining chemical relevance [43].
Approximate Algorithms: For large molecules, including proteins, approximate algorithms utilize a "direction-permutation" approach. These methods trade some computational precision for a massive reduction in processing time, making symmetry analysis feasible for biological macromolecules. Recent improvements have incorporated the Hungarian algorithm to enhance the accuracy of these approximations [43].
The standard workflow requires two primary inputs: the 3D atomic coordinates of the molecular structure and the desired symmetry point group (e.g., Cₙ, Sₙ, Cₛ, Cᵢ) against which the deviation will be measured. The output includes the CSM value, the 3D coordinates of the nearest symmetric structure, the permutation defining the symmetry operation, and the spatial direction of the symmetry element [43].
Comparative Analysis of CSM Software and Applications
The following table summarizes the key implementations and applications of continuous symmetry analysis identified in current literature.
| Software/Method Name | Core Functionality | Reported Outputs/Metrics | Key Application Contexts |
|---|---|---|---|
| CSM Software [43] | Quantifies deviation from a user-specified point group (Cₙ, Sₙ, Cₛ, Cᵢ). | CSM value (0-100), nearest symmetric structure, symmetry element direction. | Analysis of molecular distortions from dynamics, conformational changes, substitution, and reactive processes. |
| Continuous Symmetry Operation Measure [44] | Automated symmetry determination and deviation quantification for any structure describable as points in space. | Symmetry deviation yardstick, coordination geometry, molecular symmetry. | Transition metal complexes, lanthanide compounds, analysis of luminescence properties, and phase changes. |
| Symmetry Discovery via Infinitesimal Generators [45] | Discovers continuous symmetries in data beyond pre-defined affine transformations using vector fields. | Infinitesimal generators (vector fields), symmetry scores for model invariance. | Machine learning model improvement, generalization via symmetry enforcement, analysis of non-affine symmetries. |
Performance and Applicability Insights
Handling Complex Coordination Environments: The
keep-structureflag in the dedicated CSM software is critical for analyzing coordination complexes and reactive processes. It ensures the algorithm only considers permutations that maintain chemical connectivity, yielding a nearest symmetric structure that is chemically meaningful rather than just geometrically optimal [43]. This is particularly important for studying transition states or reaction pathways where bonding changes occur.Beyond Pre-Defined Symmetries: While most tools measure deviation from a pre-specified symmetry group, emerging machine learning methods aim to discover unknown symmetries directly from data. This approach uses vector fields as infinitesimal generators of continuous symmetries and is capable of identifying non-affine transformations, which are common in complex molecular systems [45].
Bridging Geometric and Electronic Structure: The connection between symmetry, coordination number, and electronic properties is a key application. For instance, density functional theory (DFT) studies on dual-atom catalysts have shown that reducing the coordination number from FeN₄CoN₄ to FeN₃CoN₃ alters the electronic structure, which in turn impacts the adsorption energy of reaction intermediates and catalytic activity [46]. CSMs provide the geometric framework to quantify such structural perturbations.
Experimental Protocols for CSM Analysis
Standard Workflow for Molecular Symmetry Quantification
The standard protocol for determining the continuous symmetry measure of a molecular structure involves a series of defined steps, as implemented in software tools [43]. The following diagram illustrates this workflow.
Title: CSM Calculation Workflow
Step-by-Step Protocol:
- Input Preparation: Obtain the 3D atomic coordinates of the molecular structure. File formats with inherent connectivity information (e.g., .mol, .mol2, .sdf, .pdb) are preferred. For formats without connectivity (e.g., .xyz), connectivity must be deduced using tools like Open Babel or provided in a separate file [43].
- Symmetry Target Selection: Specify the desired point group
Gagainst which the symmetry will be measured. The available groups typically include cyclic groups like Cₙ and Sₙ, as well as Cₛ and Cᵢ. To measure continuous chirality (CCM), the software calculates the minimal CSM with respect to all achiral point groups [43]. - Algorithm Selection:
- For small molecules, use the
exactcalculation command, ideally with thekeep-structureflag to restrict the permutation search to those that preserve chemical connectivity. This ensures the reference symmetric structure is chemically meaningful [43]. - For large molecules or proteins, use the
approx(approximate) calculation command. This method uses a direction-permutation approach to find a near-optimal solution without scanning all permutations, making the computation feasible [43].
- For small molecules, use the
- Execution and Output: Run the calculation. The primary output is the CSM value. Additional outputs include the permutation of atoms that defines the symmetry operation, the spatial direction vector of the symmetry element, and the 3D coordinates of the nearest symmetric structure that belongs to group
G[43].
Protocol for Coordination Number and Geometry Analysis
Applying CSM analysis to evaluate coordination geometry involves a more specific, chemistry-focused workflow that connects structural metrics with electronic properties.
Step-by-Step Protocol:
- Define Idealized Polyhedron: Based on the nominal coordination number from crystallographic or metric data (e.g., using Voronoi-Dirichlet partitioning), define the ideal coordination polyhedron. For example, a six-coordinate metal center often has an ideal octahedral (Oₕ) geometry, which can be measured using its rotational subgroup (e.g., C₄) [4] [44].
- Extract Coordination Sphere: From the experimental or optimized structure, isolate the central atom (e.g., a metal in a complex) and its first coordination sphere of donor atoms.
- Calculate CSM for Candidate Symmetries: Calculate the CSM value of the coordination sphere with respect to the ideal polyhedron (e.g., C₄ for octahedral). It is often informative to test multiple plausible symmetries (e.g., D₄h for square planar) to quantitatively determine the closest match [44].
- Correlate with Electronic Properties: Link the degree of geometric distortion (CSM value) to electronic or catalytic properties. For instance, in a study of Fe-Co dual-atom catalysts, a lower coordination number (FeN₃CoN₃) resulted in stronger adsorption of ORR intermediates compared to a higher coordination number (FeN₄CoN₄), directly impacting catalytic performance. The geometric distortion implied by the different coordination numbers is a key parameter that can be quantified by CSMs [46].
The following table details key software and computational resources essential for conducting research in continuous symmetry and coordination geometry analysis.
| Resource Name | Type/Format | Primary Function in Research |
|---|---|---|
| CSM Software [43] | Python-based open-source code / Online web tool | Core calculation of Continuous Symmetry and Chirality Measures for molecular structures. |
| Continuous Symmetry Operation Measure Tool [44] | Specialized software implementation | Automated determination of molecular symmetry and coordination geometry, particularly for inorganic complexes. |
| Voronoi-Dirichlet Partitioning (VDP) [4] | Algorithm in crystallographic analysis | Provides a geometric coordination number by constructing atomic domains in crystal structures, serving as an initial input for symmetry analysis. |
| Density Functional Theory (DFT) [46] | Computational quantum chemistry method | Calculates electronic structure properties (d-band center, spin polarization) to correlate with geometric distortions measured by CSMs. |
| Open Babel [43] | Chemical toolbox library | Handles file format conversion and deduces atomic connectivity for molecular structures used as input in CSM software. |
{# Gas-Phase Cluster Models for Isolating Coordination Effects
Gas-phase cluster models serve as a powerful experimental platform for isolating and studying coordination effects, providing unparalleled atomic-level insight into how the number and geometry of atoms surrounding a metal active site govern its chemical behavior. This guide compares the primary methodological approaches and their performance in determining the influence of coordination environment on catalytic properties, a critical focus for researchers developing next-generation catalysts and materials.
The Scientist's Toolkit: Essential Research Reagents and Materials
The following table details key reagents, instrumentation, and computational tools essential for research in gas-phase cluster models.
| Item Name | Function / Description |
|---|---|
| Electrospray Ionization (ESI) Source [47] | A soft ionization technique used to generate gas-phase cluster ions with intact ligands from liquid solutions, preserving specific coordination environments. |
| Linear Ion Trap Mass Spectrometer [47] | An instrument that isolates mass-selected clusters and permits the study of their reactivity with probe molecules (e.g., CO) in a controlled, gas-phase environment. |
| Cluster-Based Drag Model [48] | A computational model used in fluid dynamics simulations (CFD) to account for the effect of particle clustering on gas-solids interactions in circulating fluidized beds. |
| Trapped Ion Electron Diffraction (TIED) [49] | A hybrid experimental technique combining ion-trap mass spectrometry with electron diffraction for determining the structures of size-selected cluster ions in the gas phase. |
| Density Functional Theory (DFT) [50] [49] | A computational method used to calculate the electronic structure of atoms, molecules, and clusters. It is vital for predicting candidate structures, adsorption energies, and interpreting experimental data. |
| Generalized Coordination Number (( \overline{CN} )) [50] | An inexpensive geometric descriptor that accounts for the coordination numbers of a site's nearest neighbors, used to correlate structure with adsorption energy trends on surfaces and nanoparticles. |
Experimental Protocols for Key Methodologies
Mass Spectrometry and Gas-Phase Kinetics
This protocol, as detailed in studies of Cu-N-C model systems, is designed to probe the reactivity of clusters with defined coordination structures [47].
- Step 1: Cluster Generation and Isolation. Ligands (e.g., pyridine, bipyridine) are dissolved in methanol and co-injected with a metal salt (e.g., Cu(NO₃)₂) into an electrospray ionization (ESI) source. The desired metal-ligand complex is generated in the gas phase and then isolated based on its mass-to-charge ratio in a linear ion trap [47].
- Step 2: Reactivity Probing. A probe molecule, such as carbon monoxide (CO), is introduced into the ion trap at a known pressure to react with the isolated cluster. The reaction proceeds for a controlled time [47].
- Step 3: Product Analysis and Kinetics. The reaction products are detected by mass spectrometry. The pseudo-first-order rate constant (k) is calculated by fitting the decay of the reactant cluster signal and the growth of the product cluster signal over time, providing a quantitative measure of adsorption activity [47].
- Step 4: Electronic Structure Analysis. The experimental results are complemented with DFT calculations to determine the geometric and electronic structure of the clusters, including charge population analysis and frontier molecular orbital (FMO) characterization [47].
Trapped Ion Electron Diffraction (TIED) for Structure Determination
This method is used for experimental structure determination of cluster ions, particularly for metal clusters where computational predictions are challenging [49].
- Step 1: Beam Preparation and Size-Selection. A beam of cluster anions is produced, and a specific cluster size is selected using mass spectrometry techniques [49].
- Step 2: Electron Scattering. The size-selected cluster ensemble is trapped and exposed to a high-energy electron beam. The electrons are scattered by the atomic nuclei within the clusters [49].
- Step 3: Diffraction Pattern Collection. A detector records the scattering angles and intensities of the diffracted electrons, producing an experimental diffraction function [49].
- Step 4: Structure Assignment. The experimental diffraction pattern is compared to simulated patterns from candidate structures obtained from quantum chemical calculations (e.g., DFT). The candidate structure that best fits the experimental data is assigned as the cluster's geometry [49].
Performance Comparison of Methodologies
The table below compares the capabilities, applications, and limitations of different gas-phase cluster model approaches for studying coordination effects.
| Methodology | Key Measurable Outputs | Primary Applications | Inherent Advantages | Inherent Limitations / Challenges |
|---|---|---|---|---|
| Mass Spectrometry & Gas-Phase Kinetics [47] | Pseudo-first-order rate constant (k) for adsorbate binding; Reaction product mass spectra. | Quantifying how coordination number & geometry regulate adsorption energy & catalytic activity (e.g., CO on Cu-N-C sites). | Isolates the local coordination effect by studying clusters with atomically-precise, defined structures; Provides direct, quantitative reactivity data. | Limited to cluster sizes that can be generated and isolated; Requires careful control to avoid interference from solvent or water. |
| Trapped Ion Electron Diffraction (TIED) [49] | Experimental diffraction function; Assigned 3D atomic structure (bond lengths, angles). | Determining the ground-state and isomeric structures of metal clusters (e.g., Ptn-, Run-, Bin-). | Provides direct, experimental determination of cluster geometry, serving as a benchmark for theory. | Experimentally demanding; Structure assignment relies on the quality and availability of theoretical candidate structures. |
| DFT + Geometric Descriptor (e.g., GCN) [50] | Generalized Coordination Number (( \overline{CN} )); Predicted adsorption energies (ΔEads); d-band center. | Establishing structure-energy correlations; Creating catalytic activity plots and selectivity maps for extended surfaces & nanoparticles. | Inexpensive to compute; Provides a simple, intuitive link between geometry and reactivity; Enables atom-by-atom catalyst design. | A purely theoretical descriptor; Accuracy depends on the exchange-correlation functional; Assumes no significant surface reconstruction upon adsorption. |
| Cluster-Based Drag Models (CFD) [48] | Cluster phase fraction; Average solids holdup; Cluster size and velocity distributions. | Studying hydrodynamic clustering of particles in gas-solids circulating fluidized bed (CFB) risers. | Models macroscopic clustering behavior in industrial processes; Can be validated against reactor data. | Focused on fluid dynamics of particle aggregates, not atomic-level coordination chemistry. |
Quantitative Insights from Gas-Phase Studies
Experimental data from gas-phase studies provides direct evidence of how coordination number dictates reactivity.
- Coordination Number vs. CO Adsorption on Cu(I) Centers: A study on defined Cu(I)-pyridinic N clusters revealed that the CO adsorption rate constant (k) and bonding strength are non-linearly dependent on the N coordination number. [Cu-bpy]+ (two N) showed higher activity than [Cu-Py]+ (one N), while [Cu-tpy]+ (three N) was largely inert. The adsorption enthalpy for [Cu-Py]+ was measured to be -138.02 kJ mol⁻¹ [47].
- Orbital Moments in Cobalt Clusters: Investigations of deposited Co clusters on platinum surfaces demonstrated that decreased coordination number leads to significantly enhanced orbital magnetic moments. Small Co nanoparticles exhibited orbital moments up to 1.1 μB per atom, which decreases toward the bulk value as cluster size increases and coordination numbers rise [51].
- Generalized Coordination and Reactivity: The Generalized Coordination Number (( \overline{CN} )) has been successfully correlated with adsorption energies for key intermediates like *OH, *OOH, and *CO on transition metal surfaces. This geometric descriptor can predict trends in catalytic activity for reactions such as the oxygen reduction reaction (ORR) [50].
Workflow for Isolating Coordination Effects Using Gas-Phase Cluster Models
Gas-phase cluster models provide an indispensable, orthogonal approach to traditional surface science for deconvoluting the complex interplay between coordination environment and chemical function. While mass spectrometry and TIED offer direct experimental observation of reactivity and structure, computational descriptors like GCN provide a powerful framework for generalization and prediction. The integration of these methods, as illustrated in the workflow, creates a robust pipeline for achieving an atomic-level understanding, ultimately guiding the rational design of catalysts with tailored coordination sites for enhanced selectivity and activity in applications from drug development to energy conversion.
Computational methods have become indispensable tools in modern scientific research, providing atomistic insights into material and biomolecular systems that are often challenging to obtain experimentally. Among these methods, Density Functional Theory (DFT) and Ab Initio Molecular Dynamics (AIMD) simulations represent two cornerstone approaches for investigating structural, electronic, and dynamic properties from first principles. While both methods aim to provide accurate quantum mechanical descriptions of atomic-scale behavior, they differ significantly in their computational approaches, applicability, and performance characteristics. DFT focuses primarily on solving for the ground-state electronic structure, enabling the prediction of physical and chemical properties based on electron density distributions. In contrast, AIMD simulations extend beyond static calculations to model the time evolution of atomic positions while explicitly accounting for electronic structure effects at each step. This comparison guide objectively evaluates the performance characteristics, accuracy, and practical implementation of these computational approaches within the specific context of coordination number determination and biomolecular applications, providing researchers with a comprehensive framework for selecting appropriate methodologies for their specific scientific inquiries.
Theoretical Foundations and Methodological Approaches
Density Functional Theory (DFT) Fundamentals
DFT operates on the fundamental principle that the ground-state energy of a quantum mechanical system can be expressed as a functional of the electron density, rather than requiring the complex many-electron wavefunction. This theoretical foundation dramatically reduces the computational complexity compared to wavefunction-based methods, making it feasible to study systems containing hundreds to thousands of atoms. Modern DFT implementations employ various approximations for the exchange-correlation functional, with the generalized gradient approximation (GGA) being among the most widely used. For example, in the study of Half Heusler FeAsNb alloys for electrothermal actuators, researchers utilized the GGA-WC functional to determine structural, elastic, and transport properties [52]. The FP-LAPW (full-potential linearized augmented-plane wave) method as implemented in the Wien2K code provides an all-electron approach that does not approximate the potential shape, offering high accuracy for solid-state systems. DFT's ability to predict diverse material properties—from electronic band structures to thermal conductivity using the Slack formula—makes it particularly valuable for materials design and characterization, though its computational scaling of approximately O(N³) with system size presents limitations for large biomolecular systems [52] [53].
Ab Initio Molecular Dynamics Methodologies
AIMD simulations integrate the accuracy of quantum mechanical calculations with the dynamic sampling capabilities of molecular dynamics. Traditional AIMD employs DFT at each dynamics step to compute forces acting on atoms, enabling trajectory evolution with quantum mechanical accuracy. However, the computational expense of this approach—with similar O(N³) scaling as DFT—has limited its application to relatively small systems and short timescales. Recent breakthroughs have addressed this limitation through innovative machine learning approaches. The AI2BMD system exemplifies this advancement, combining a protein fragmentation scheme with machine learning force fields to achieve ab initio accuracy for proteins exceeding 10,000 atoms [53]. This methodology fragments proteins into manageable dipeptide units (typically 12-36 atoms), calculates intra- and inter-unit interactions using MLFFs trained on DFT data, and assembles the results to determine total protein energy and forces. By leveraging physics-informed molecular representations and efficient algorithms with linear time complexity, AI2BMD maintains quantum mechanical accuracy while reducing computational time by several orders of magnitude compared to conventional DFT-based AIMD [53].
Table 1: Fundamental Methodological Comparisons Between DFT and Ab Initio MD
| Feature | Density Functional Theory (DFT) | Ab Initio Molecular Dynamics (AIMD) |
|---|---|---|
| Primary Focus | Electronic structure calculation | Time evolution of atomic positions |
| Theoretical Basis | Hohenberg-Kohn theorems [4] | Born-Oppenheimer or Car-Parrinello dynamics |
| Key Approximation | Exchange-correlation functional | Potential energy surface representation |
| System Size Limitation | ~100-1,000 atoms [53] | ~10-1,000 atoms (traditional), >10,000 atoms (AI2BMD) [53] |
| Time Scale Limitation | Static calculations only | Femtoseconds to nanoseconds [53] |
| Computational Scaling | O(N³) [53] | O(N³) to O(N) depending on implementation [53] |
Performance Comparison: Accuracy and Computational Efficiency
Accuracy Assessment for Biomolecular Systems
Rigorous validation against experimental data and high-level theoretical references reveals significant differences in the accuracy profiles of DFT and AIMD methods. For protein systems, the AI2BMD approach demonstrates remarkable accuracy in energy and force calculations when benchmarked against DFT references. In evaluations across multiple proteins ranging from 175 to 13,728 atoms, AI2BMD achieved a mean absolute error (MAE) of just 0.038 kcal mol⁻¹ per atom for potential energy calculations, outperforming conventional molecular mechanics force fields by approximately two orders of magnitude [53]. For atomic forces, AI2BMD maintained an MAE of 1.974 kcal mol⁻¹ Å⁻¹ compared to DFT references, significantly lower than the 8.094 kcal mol⁻¹ Å⁻¹ observed with molecular mechanics approaches [53]. This accuracy extends to the prediction of experimental observables such as 3J couplings from nuclear magnetic resonance experiments, with AI2BMD successfully reproducing protein folding and unfolding processes that match experimental data [53].
Traditional AIMD implementations relying directly on DFT calculations, while highly accurate, face severe limitations when applied to biomolecular systems due to their computational expense. For a medium-sized protein like Trp-cage (281 atoms), a single DFT-based dynamics step requires approximately 21 minutes of computation time, making nanosecond-scale simulations—requiring billions of steps—computationally prohibitive [53]. This limitation fundamentally restricts the ability of conventional DFT-based AIMD to sample biologically relevant timescales for protein folding and conformational changes.
Computational Efficiency and Scaling Behavior
The computational efficiency disparity between standard DFT calculations and advanced AIMD implementations becomes particularly evident as system size increases. While both methods formally exhibit O(N³) scaling in their traditional implementations, machine learning approaches like AI2BMD achieve near-linear scaling through architectural innovations [53]. This dramatic improvement in computational efficiency enables simulations that would be impossible with conventional DFT. For the albumin-binding domain (746 atoms), AI2BMD requires just 0.125 seconds per simulation step compared to 92 minutes for DFT—an improvement of nearly five orders of magnitude [53]. This efficiency advantage expands further for larger systems; for aminopeptidase N (13,728 atoms), AI2BMD completes a simulation step in 2.610 seconds, while DFT would require an estimated 254 days, representing a computational speedup exceeding six orders of magnitude [53].
Table 2: Computational Performance Comparison for Protein Systems
| Protein System | Number of Atoms | DF Computation Time | AI2BMD Computation Time | Speedup Factor |
|---|---|---|---|---|
| Trp-cage | 281 | 21 minutes/step [53] | 0.072 seconds/step [53] | ~17,500× |
| Albumin-binding Domain | 746 | 92 minutes/step [53] | 0.125 seconds/step [53] | ~44,160× |
| Aminopeptidase N | 13,728 | ~254 days/step [53] | 2.610 seconds/step [53] | ~8,400,000× |
These efficiency gains do not come at the expense of accuracy when properly validated. The AI2BMD system demonstrates robust performance across diverse protein conformations—folded, unfolded, and intermediate states—with minimal fluctuation in accuracy metrics [53]. This combination of speed and precision enables previously infeasible simulations, including precise free-energy calculations for protein folding and the estimation of thermodynamic properties that align closely with experimental measurements [53].
Experimental Protocols and Validation Methodologies
DFT Validation for Material Property Prediction
The protocol for validating DFT calculations of material properties follows a systematic approach combining theoretical predictions with experimental verification. In the analysis of Half Heusler FeAsNb alloys for electrothermal actuators, researchers employed the following methodology: First, initial crystal structures were optimized using the FP-LAPW method within the Wien2K code, employing GGA-WC exchange-correlation functionals [52]. Subsequently, elastic constants were derived from strain-derivative calculations, followed by the determination of mechanical properties including bulk modulus, shear modulus, and Young's modulus. Electronic properties were calculated via band structure and density of states analyses, while transport properties including electrical conductivity and Seebeck coefficient were obtained using the BoltzTraP code [52]. Thermal properties, particularly lattice thermal conductivity, were evaluated using the Slack formula, which considers phonon scattering processes. Finally, these calculated properties informed finite element analysis of electrothermal actuator performance, comparing the displacement response under applied voltage for FeAsNb against traditional polysilicon materials [52]. This comprehensive protocol enables confident prediction of material performance prior to synthesis and experimental characterization.
AIMD Validation for Biomolecular Simulations
Validating the accuracy of AIMD simulations requires comparison against both quantum mechanical benchmarks and experimental data. The AI2BMD validation protocol exemplifies rigorous methodology: First, a comprehensive training dataset was constructed by scanning main-chain dihedrals of all protein units and running AIMD simulations with the 6-31g* basis set and M06-2X functional, generating 20.88 million samples [53]. The machine learning force field was then trained on this dataset using the ViSNet architecture, which incorporates physics-informed molecular representations and calculates four-body interactions with linear time complexity. For validation, simulations were performed for nine proteins with sizes ranging from 175 to 13,728 atoms, each initiated from five folded, five unfolded, and ten intermediate structures derived from replica-exchange MD simulations [53]. The resulting energy and force calculations were compared directly against DFT references where feasible, while larger systems employed fragmented DFT calculations as benchmarks. Additional validation involved comparing simulation-derived 3J couplings with nuclear magnetic resonance experimental data and assessing protein folding thermodynamics against experimental melting temperatures [53]. This multi-faceted validation protocol ensures both quantum mechanical accuracy and biological relevance of the simulation results.
Figure 1: AI2BMD Simulation Workflow for Protein Dynamics
Coordination Number Determination in Structural Analysis
Traditional Approaches and Limitations
The accurate determination of coordination numbers represents a fundamental challenge in structural analysis across chemistry and materials science. Traditional methods for coordination number evaluation primarily rely on geometric considerations based on atomic positions. The distance-based method analyzes sequences of interatomic distances, defining the first coordination sphere by either a fixed cut-off value (dmin√2) or a tolerance-based approach (typically 15% between dmin and dmax) [4]. The Brunner-Schwarzenbach method identifies the first large gap in the distance sequence, though this approach often suffers from coordination reciprocity issues where atom A may be counted in the coordination sphere of B, but not vice versa [4]. Voronoi-Dirichlet partitioning offers an alternative geometric approach that constructs spatial domains around each atom, considering atoms as coordinated if their Voronoi polyhedra share a common face [4]. While these methods provide valuable structural insights, they exhibit limitations in complex systems, particularly for intermetallic compounds where bonding mechanisms are not well established. The inherent challenge lies in condensing complete geometrical information about spatial atomic arrangements into a single number that accurately represents chemical coordination.
Electron Density-Based Approaches
Modern approaches to coordination number determination increasingly leverage electron density distributions from DFT calculations or experimental measurements, providing a more physically grounded foundation for coordination analysis. The topological coordination number approach utilizes quantum theory of atoms in molecules (QTAIM) interatomic surfaces to calculate solid angles subtended at nuclear positions by each diatomic contact surface [4]. This method represents a significant advancement over purely geometric approaches by naturally incorporating the effects of different atomic sizes and electron-density decay, even in highly symmetrical crystal structures [4]. Unlike Voronoi-Dirichlet partitioning, which typically yields coordination numbers larger than chemically expected values, the topological approach enables the identification of chemically meaningful coordination numbers through coordination reciprocity requirements. This method excels particularly in complex structures such as intermetallic phases, where it can numerically rank different sub-coordination scenarios of similar importance, providing a more nuanced characterization that includes relative weights and associated effective coordination numbers [4]. Since electron density distribution represents a fundamental quantum mechanical observable according to the Hohenberg-Kohn theorems, coordination numbers derived from this physical property offer enhanced predictive power for structure-property relationships in materials design [4].
Figure 2: Electron Density-Based Coordination Analysis Workflow
Research Reagent Solutions: Computational Tools and Materials
Table 3: Essential Computational Tools for DFT and Ab Initio MD Simulations
| Tool/Code | Primary Function | Application Domain | Key Features |
|---|---|---|---|
| Wien2K | FP-LAPW DFT calculations [52] | Solid-state materials | All-electron, full-potential method for high-precision electronic structure |
| BoltzTraP | Transport property calculation [52] | Thermoelectric materials | Boltzmann transport theory implementation for conductivity and Seebeck coefficient |
| Gibbs Code | Thermal property determination [52] | Materials characterization | Thermodynamic properties from DFT calculations |
| AI2BMD | Ab initio biomolecular dynamics [53] | Protein folding and dynamics | MLFF with protein fragmentation for ab initio accuracy on large systems |
| AMOEBA | Polarizable force field [53] | Solvent modeling | Explicit polarizable solvent for biomolecular simulations |
| ViSNet | Machine learning force field [53] | Molecular simulations | Physics-informed representations with linear time complexity |
This comprehensive comparison demonstrates that both DFT calculations and ab initio MD simulations offer powerful capabilities for investigating atomic-scale phenomena, though with distinct strengths and optimal application domains. DFT excels in predicting electronic structure and material properties for systems of moderate size, with established protocols for validation against experimental measurements. The method has proven particularly valuable for materials design, as evidenced by its successful application in characterizing Half Heusler alloys for electrothermal actuators [52]. Meanwhile, ab initio MD simulations, particularly next-generation implementations like AI2BMD, enable the investigation of biomolecular dynamics with quantum mechanical accuracy across biologically relevant timescales and system sizes [53]. These advanced approaches achieve computational speedups exceeding six orders of magnitude compared to conventional DFT while maintaining ab initio accuracy in energy and force calculations [53]. For coordination number determination, electron density-based topological approaches provide more chemically meaningful descriptions than traditional geometric methods, particularly for complex intermetallic compounds [4]. As both computational methodologies continue to evolve, their synergistic application promises to expand the frontiers of predictive materials design and biomolecular simulation, enabling researchers to address increasingly complex scientific challenges across chemistry, materials science, and drug development.
Addressing Methodological Limitations and Optimizing Accuracy
Resolving Coordination Reciprocity Violations in Traditional Methods
In the analysis of crystal structures, the coordination number (CN) is a fundamental metric, condensing complex geometrical information about a central atom's environment into a single integer. Traditionally defined as the number of atoms in the first coordination sphere, the CN is vital for understanding material properties and bonding behavior [15]. However, the conventional methods for determining CNs, particularly in complex solids like intermetallic compounds, suffer from a significant flaw: coordination reciprocity violations [15].
A reciprocity violation occurs when, for the same interatomic distance between atoms A and B, atom A is counted in the coordination sphere of B, but the reverse is not true. This lack of pairwise symmetry is chemically implausible from a physical chemistry perspective, as bonding interactions are inherently symmetric [15]. Such inconsistencies prevent a coherent description of crystal structures and complicate the integration of bonding information, such as energy calculations [15]. This guide objectively compares the performance of traditional methods against a modern topological approach that successfully resolves these violations, providing researchers with a framework for selecting accurate characterization tools.
Traditional Methods and Their Limitations
Traditional CN determination methods rely primarily on geometric information derived from crystallographic data but differ in their specific algorithms.
Distance-Based String Methods
The simplest approaches analyze the sequence of interatomic distances from a central atom.
- Principle: The first coordination sphere is identified by a cut-off value, often based on the minimal distance (e.g., dmin√2) or by allowing a tolerance (e.g., 15%) between the shortest and longest distances within the sphere [15].
- The Brunner-Schwarzenbach (BS) Method: This is a more sophisticated gap-based method used in major databases like Pearson's Crystal Data. It identifies the first large gap in the sequence of increasing interatomic distances to define the coordination sphere [15].
Voronoi-Dirichlet Partitioning (VDP)
This geometric construction defines an atom's domain as the region of space closer to it than to any other atom.
- Principle: Atoms are considered coordinated if their Voronoi polyhedra share a face [15]. The number of faces provides the maximum possible CN, which is often later reduced to identify the chemically meaningful coordination [15].
Table 1: Limitations of Traditional Coordination Number Determination Methods
| Method | Core Principle | Key Limitations |
|---|---|---|
| Distance-Based (Brunner-Schwarzenbach) | Identifies the first large gap in a sequence of interatomic distances [15]. | - Requires an arbitrary cut-off parameter [15].- Highly susceptible to coordination reciprocity violations [15].- More than one gap of similar size can lead to ambiguous results (e.g., α-Fe with CN=6+8) [15]. |
| Voronoi-Dirichlet Partitioning (VDP) | Atoms are coordinated if their Voronoi polyhedra share a face [15]. | - Provides a maximum CN value that is often too high chemically [15].- Requires subsequent, often biased, methods to distinguish "direct" from "indirect" neighbors [15]. |
A Modern Solution: The Topological Coordination Number (tCN) Approach
The topological coordination number (tCN) method represents a paradigm shift by basing its analysis on the experimentally accessible electron density (ED) rather than pure geometry, directly addressing the shortcomings of traditional techniques [15].
Theoretical Foundation
The tCN approach is rooted in the Quantum Theory of Atoms in Molecules (QTAIM). It uses triangulated surface data sets of QTAIM interatomic surfaces to calculate the solid angles subtended at the nuclear positions by each diatomic contact surface [15]. This process generalizes the VDP-based procedure by naturally incorporating the effect of different atomic sizes through their electron-density distributions [15].
Resolving Reciprocity
The core strength of the tCN approach is its built-in coordination reciprocity requirement. The method ranks different sub-coordination scenarios using weighting functions derived from geometrical properties, ensuring that the final identification of chemically meaningful coordination numbers is consistent and pairwise symmetric [15]. This eliminates the physically implausible violations inherent in BS-type methods.
Performance Comparison: Traditional vs. Topological Methods
The following comparative data highlights the practical differences in output between these methods.
Table 2: Comparative Analysis of Traditional vs. Topological CN Methods
| Performance Metric | Traditional (VDP/Distance-Based) | Topological (tCN) Approach |
|---|---|---|
| Theoretical Basis | Purely geometrical (atomic positions) [15]. | Electron-density distribution (QTAIM) [15]. |
| Coordination Reciprocity | Often violated, leading to non-symmetric coordination [15]. | Enforced by the methodology, ensuring pairwise symmetry [15]. |
| Effect of Atomic Size | Often requires manual correction or is ignored. | Naturally included via the electron-density decay [15]. |
| Result for Highly Symmetric Element Structures | Provides a single, integer CN. | Can reveal differences even in high-symmetry structures due to electron-density effects [15]. |
| Result for Complex Structures (e.g., TiNiSi type) | Often forces a single, sometimes arbitrary, CN choice. | Provides a ranked list of different coordination scenarios with their relative weights and effective CNs, offering a more nuanced characterization [15]. |
| Suitability for AI/ML | Problematic due to inherent inconsistencies (e.g., reciprocity violations). | Highly suitable as it provides a digitizable, consistent topological description for graph neural networks [15]. |
Experimental Protocols for Coordination Number Determination
Protocol for Molecular Dynamics-Based CN Calculation
This protocol is commonly used for studying solvation structures, as in the investigation of bivalent ions in organic solvents [54].
- System Setup: Create a simulation box containing the solute (e.g., an ion) and several hundred solvent molecules. Energy minimization is performed to preclude atomic overlaps [54].
- Equilibration: The system is brought to equilibrium at the target temperature and pressure (e.g., 330 K, 1 bar) using a thermostat and barostat within a molecular dynamics package like GROMACS [54].
- Production Run: A sufficiently long simulation (e.g., 1.55 ns) is performed to collect trajectory data. Results are averaged over time and multiple independent trajectories [54].
- Radial Distribution Function (RDF) Calculation: The RDF, gab(r), between the solute and solvent atoms is computed from the trajectory data [55].
- CN Integration: The coordination number is calculated by integrating the RDF up to the position of the first minimum, rmin, using the formula: CN(r) = 4πρ ∫0r gab(r)r² dr where ρ is the number density of the solvent [55].
Protocol for Topological CN (tCN) Analysis from Crystallographic Data
This protocol outlines the steps for the tCN method based on electron density [15].
- Electron Density Source: Obtain an experimental or theoretical electron-density distribution for the crystal structure under investigation.
- QTAIM Analysis: Perform a topological analysis of the electron density according to QTAIM to identify atomic basins and interatomic surfaces.
- Solid Angle Calculation: For each diatomic contact, use triangulated surface data sets of the QTAIM interatomic surfaces to calculate the solid angle subtended at the nuclear position.
- Sub-Coordination Scenario Ranking: Apply weighting functions (derived from geometrical properties of square and semicircle areas) to rank different possible sub-coordination scenarios.
- Effective tCN Evaluation: Calculate the effective topological coordination number by summing the weights of the neighbors in the chemically meaningful coordination sphere, ensuring coordination reciprocity.
The following workflow diagram illustrates the logical sequence and comparative outputs of these protocols.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Computational Tools for Coordination Number Research
| Item / Software | Function in Research |
|---|---|
| GROMACS [54] | A software toolkit for performing molecular dynamics simulations, used to model ion-solvent interactions and calculate RDFs. |
| Molecular Dynamics Force Fields (e.g., GROMOS) [54] | Parametrized potentials describing interatomic interactions; vital for accurate simulation of solvation shells. |
| Spatial Light Modulator (SLM) [56] | A device used in advanced microscopy (e.g., SIM) to generate high-contrast illumination patterns for super-resolution imaging of cellular structures. |
| Cambridge Structural Database (CSD) [55] | A repository of experimentally determined organic and metal-organic crystal structures used for statistical analysis of coordination geometries. |
| Automated Topology Builder (ATB) [54] | A database providing molecular topology and partial atomic charges for solvent and solute molecules used in molecular dynamics simulations. |
| Quantum Theory of Atoms in Molecules (QTAIM) [15] | A theoretical framework for analyzing electron density distributions to define atomic boundaries and bond paths, forming the basis for the tCN approach. |
| Pearson's Crystal Data [15] | A database of inorganic crystal structures that utilizes the Brunner-Schwarzenbach method for coordination number analysis. |
Handling Dynamic and Heterogeneous Coordination Environments
In molecular sciences and materials engineering, the coordination environment of a metal center—defined by the number, type, and spatial arrangement of its immediate neighbors—is a critical determinant of functional properties. While classical analysis often treats these environments as static, real-world systems in catalysis, drug discovery, and energy materials frequently exhibit dynamic and heterogeneous coordination. This complexity arises from fluctuating conditions, diverse structural motifs, and dynamic binding equilibria, presenting significant challenges for accurate characterization and functional prediction.
This guide provides a comparative evaluation of modern methodologies developed to address these challenges. It contrasts traditional geometric approaches with advanced techniques rooted in electron density analysis, spectroscopic correlation, and templated synthesis. The focus is on quantifying the accuracy and applicability of these methods across diverse systems—from single-atom catalysts to complex intermetallic compounds—providing researchers with a structured framework for selecting appropriate characterization strategies in their work.
Comparative Analysis of Methodologies and Performance Data
The following analysis compares the core principles, advantages, and limitations of prominent methods for investigating coordination environments. Quantitative performance data, where available, are summarized to facilitate objective comparison.
Table 1: Method Comparison for Coordination Environment Analysis
| Method | Core Principle | Typical Application | Key Performance Metrics | Notable Advantages |
|---|---|---|---|---|
| Topological Coordination Number (tCN) [4] | Analysis of electron density via QTAIM to calculate solid angles subtended by interatomic surfaces. | Intermetallic phases, complex inorganic crystals (e.g., TiNiSi type). | Quantifies relative weights of different sub-coordination scenarios; accounts for atomic size effects. | Provides coordination-consistent, chemically meaningful numbers; inherently includes atomic size effects. |
| Multi-Modal Spectroscopy & MCR-ALS [8] | Correlation of XAS and optical spectroscopy data using Multivariate Curve Resolution to deconvolute coexisting states. | Speciation of metal ions in dynamic systems like molten salts (e.g., Ni(II) in LiCl–KCl). | Identifies number of unique coordination states and their mixing fractions as a function of temperature. | Resolves dynamic heterogeneity without prior knowledge of standards; provides population distributions. |
| Gas-Phase Cluster Modeling [57] | Electrospray ionization mass spectrometry to generate and react precisely defined coordination complexes. | Decoupling individual coordination effects in Single-Atom Catalysts (e.g., Cu–N–C for CO adsorption). | Pseudo-first-order rate constants (k) for reactions like CO adsorption. | Provides atomic-level, unambiguous structure-activity relationships free from support heterogeneity. |
| NaCl Templated Synthesis [58] | Use of recyclable NaCl template to control coordination geometry during high-temperature pyrolysis. | Scalable production of tailored Single-Atom Catalysts (e.g., Cl–Fe–N4 sites). | Mass yield of SACs: 18.3% - 50.9%; Coordination number control (e.g., Fe–N: 4.3, Fe–Cl: 1.0). | Achieves precise, tailored coordination environments (e.g., axial M–Cl bonds) with high mass yield. |
| DFT Analysis of Coordination Number [46] | Density Functional Theory calculations to model electronic structure and reaction pathways. | Rational design of catalyst active sites (e.g., FeN3CoN3 vs. FeN4CoN4 for ORR). | Free energy of reaction steps (ΔGO, ΔGOH); d-band center. | Uncovers electronic origins of activity; predicts performance prior to synthesis. |
Table 2: Quantitative Performance Data from Key Studies
| Study System | Method | Key Quantitative Finding | Implication for Accuracy |
|---|---|---|---|
| Cu–N–C Clusters [57] | Gas-Phase Kinetics | Rate constant (k) for [Cu-bpy]+ CO adsorption: ~2.5x higher than [Cu-Py]+; [Cu-tpy]+ inert. | Directly quantifies how N-coordination number modulates reactivity, excluding surface effects. |
| FeN3CoN3 vs. FeN4CoN4 [46] | DFT | Limiting potential for *OH removal: 0.36 eV (FeN3CoN3) vs. 0.28 eV (FeN4CoN4). | 4N-coordination lowers the overpotential, identifying it as the more effective catalytic model. |
| Ni(II) in LiCl–KCl [8] | XAS/MCR-ALS | Identified 2-3 distinct coordination states of Ni(II) with temperature-dependent mixing fractions. | Accurately captures heterogeneous speciation that conventional EXAFS fitting would average out. |
| Fe1CNCl SAC [58] | EXAFS Fitting | Coordination numbers: Fe–N = 4.3, Fe–Cl = 1.0. Bond distances: Fe–N = 1.91 Å, Fe–Cl = 2.26 Å. | Confirms successful synthesis of a precisely tailored penta-coordinate Cl–Fe–N4 site. |
Experimental Protocols for Key Methodologies
This protocol determines coordination numbers from electron density distributions, providing a more chemically meaningful alternative to purely geometric methods.
1. Electron Density Calculation:
- Obtain the experimental or theoretical electron density distribution of the crystal structure.
- Source Requirement: High-quality diffraction data or a converged quantum chemical calculation is essential.
2. Identification of Interatomic Surfaces:
- Perform a Quantum Theory of Atoms in Molecules (QTAIM) analysis.
- Calculate the interatomic surfaces between the central atom and all its potential neighbors.
3. Solid Angle Calculation:
- Use triangulated surface data sets of the QTAIM interatomic surfaces.
- Compute the solid angle subtended at the central nuclear position by each diatomic contact surface.
4. Coordination Ranking and Reciprocity:
- Rank different sub-coordination scenarios using weighting functions derived from geometrical properties.
- Apply coordination reciprocity requirements to extract a coordination-consistent final number.
Key Data Interpretation: The result is often not a single integer but a listing of different chemically plausible coordination scenarios with their associated relative weights and effective coordination numbers. This is particularly useful for complex structures like intermetallic phases.
This protocol resolves multiple, coexisting coordination states of a metal ion in a dynamic environment like a molten salt.
1. Synchrotron XAS Data Collection:
- Prepare a sample with the metal solute (e.g., 1 wt% NiCl₂) in the solvent matrix (e.g., LiCl–KCl eutectic).
- Use a custom high-temperature cell for in situ XAS data acquisition across a temperature range (e.g., from solid to molten state).
- Collect data at a high signal-to-noise ratio, typically at a synchrotron beamline.
2. Optical Absorption Spectroscopy:
- Collect complementary UV-Vis-NIR spectra on the same or a chemically identical sample over the same temperature range.
- Use a lower solute concentration (e.g., 0.1 wt%) to ensure spectra are within a valid absorbance range.
3. Multivariate Curve Resolution - Alternating Least Squares (MCR-ALS) Analysis:
- Perform Principal Component Analysis (PCA) on the XANES spectral series to determine the number of independent components (unique coordination states).
- Input this number into the MCR-ALS algorithm to iteratively separate the spectral components and their concentration profiles across temperatures without requiring pre-defined standards.
4. Structural Assignment via AIMD:
- Use Ab Initio Molecular Dynamics (AIMD) simulations of the Ni(II) ion in the molten salt to generate model coordination environments.
- Compare the theoretical XANES spectra calculated from AIMD trajectories to the MCR-ALS deconvoluted components for final structural assignment.
Key Data Interpretation: The output provides the distinct XANES spectra for each coordination state and their mixing fractions (concentrations) at every measured temperature, directly quantifying the dynamic heterogeneity.
This protocol isolates the effect of coordination number by studying mass-selected clusters, removing support and heterogeneity effects.
1. Cluster Generation and Mass Selection:
- Dissolve metal salt (e.g., Cu(NO₃)₂) and desired ligand (e.g., pyridine, bipyridine) in methanol.
- Introduce the solution into an electrospray ionization (ESI) mass spectrometer.
- Use collision-induced dissociation (CID) or direct isolation to generate and select the desired Cu(I)-ligand complex (e.g., [Cu(bpy)]⁺).
2. Gas-Phase Reaction Kinetics:
- Introduce a controlled pressure of a probe molecule (e.g., CO) into the ion trap containing the mass-selected clusters.
- Allow the clusters and gas to react for a defined period (e.g., 30 ms).
- Detect the reaction products using mass spectrometry.
3. Rate Constant Calculation:
- Monitor the decay of the reactant cluster peak and the growth of the product peak (e.g., [Cu(bpy)(CO)]⁺) over different reaction times.
- Fit the data to a pseudo-first-order kinetic model to extract the rate constant (k) for the ligand adsorption reaction.
Key Data Interpretation: The rate constants for different clusters (e.g., [Cu(Py)]⁺ vs. [Cu(bpy)]⁺) provide a direct, quantitative comparison of the intrinsic reactivity imparted by specific coordination environments.
Visualization of Workflows and Coordination Pathways
The following diagrams illustrate the logical flow of the key experimental and analytical processes described in this guide.
Multi-Modal Speciation Analysis
Coordination Impact on Catalytic Pathway
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Materials
| Item | Function in Research | Specific Application Example |
|---|---|---|
| NaCl Template [58] | A recyclable, low-cost hard template to direct 3D morphology and control axial coordination (M–Cl) during high-temperature pyrolysis. | Scalable synthesis of single-atom catalysts with Cl–M–N4 coordination. |
| Dicyandiamide [58] | A common nitrogen source used in the pyrolysis synthesis of nitrogen-doped carbon supports for anchoring metal atoms. | Creating N-doped carbon frameworks in SACs and DACs. |
| Synchrotron-Grade XAS Cell [8] | A custom-built, high-temperature cell for in situ X-ray absorption spectroscopy measurements under controlled atmosphere. | Studying coordination states of metal ions in corrosive molten salts. |
| Electrospray Ionization Source [57] | A soft ionization technique to generate gas-phase ions of intact metal-ligand complexes from solution for mass spectrometry. | Producing defined [CuL]+ clusters for gas-phase reactivity studies. |
| Linear Ion Trap Mass Spectrometer [57] | An instrument for isolating specific ion masses and studying their gas-phase reactions with probe molecules. | Measuring kinetics of CO adsorption on mass-selected Cu coordination complexes. |
In coordination chemistry, the strategic design of complex ligands is fundamental to controlling the structure and function of metal complexes. Ligands are ions or molecules that bind to a central metal atom to form a coordination complex, and their binding strategies—including chelating, ambidentate, and polydentate systems—directly influence properties such as stability, reactivity, and electronic characteristics. These strategies are crucial for applications ranging from catalysis and materials science to pharmaceutical development. Understanding the relationships between ligand architecture and metal complex performance enables researchers to tailor materials with precision. This guide objectively compares the performance of different ligand strategies, drawing on experimental data to highlight their distinct advantages and limitations within the broader context of evaluating coordination number determination methods.
Comparative Performance of Ligand Systems
The table below summarizes key performance characteristics of different ligand types, based on experimental data from recent studies. This comparison highlights how ligand architecture influences stability, selectivity, and application-specific efficacy.
Table 1: Comparative Performance of Ligand Systems in Metal Complexation
| Ligand System / Property | Coordination Number / Mode | Experimental Performance / Observed Effect | Key Application Context |
|---|---|---|---|
| Phosphonic Acid-Based Polydentate [59] | Varies from monodentate (H(3)PO(4)) to pentadentate (DTPMP) | Optimal Performance with DTPMP: Superior suspension stability, material removal rate (MRR), and surface quality in Chemical Mechanical Polishing (CMP). | Development of high-performance CeO(_2) polishing slurries for semiconductors. |
| Ambidentate Mercaptocarboxylates [60] | S-monodentate, S,O-chelating, or S-bridging | Variable Binding Modes: Ligands like MTA and MPA show different coordination (monodentate, chelating, bridging) depending on the metal and ancillary ligands. | Synthesis of Platinum Group Metal (PGM) complexes with tunable structures. |
| Iminophosphonamide Chelators [61] | Primarily N,N'-bidentate chelation | Versatile Scaffold: Forms stable complexes across s-, p-, d-, and f-block metals; steric and electronic properties tunable via substituents. | Synthetic inorganic chemistry; creation of non-metallocene complexes. |
| Polydentate N/C Ligands [62] | Tetradentate (CCCN) to Pentadentate (CCCCN, NCCCN) | Planar High-Coordinate Complexes: Successive modification of coordinating atoms allows access to rare planar penta- and hexacoordinate geometries. | Synthesis of organometallic species with photoacoustic and photothermal properties. |
Experimental Protocols and Methodologies
To ensure reproducibility and provide a clear basis for the performance data cited, this section details the key experimental methodologies from the referenced studies.
This protocol outlines the procedure for modifying cerium oxide (CeO(_2)) particles with phosphonic acid ligands and evaluating their performance in chemical mechanical polishing (CMP).
Key Research Reagents:
- Abrasives: CeO(_2) nanoparticles synthesized from cerium carbonate.
- Ligands: H(3)PO(4) (monodentate), ATMP (tridentate), DTPMP (pentadentate).
- Equipment: Nanoscale grinder, density functional theory (DFT) computational setup.
Experimental Workflow:
- Particle Synthesis: Calcinate cerium carbonate (Ce(2)(CO(3))(3)·6H(2)O) at 600 °C for 12 hours to obtain CeO(_2).
- Grinding: Process the calcined product using a nanoscale grinder to produce CeO(_2) nanoparticles.
- Surface Modification: Disperse CeO(2) nanoparticles in deionized water. Add a specific ligand (H(3)PO(_4), ATMP, or DTPMP) and heat the mixture at 100 °C for 12 hours to facilitate surface modification.
- Performance Testing:
- Suspension Stability: Measure the stability of the modified slurries over time.
- Material Removal Rate (MRR): Perform CMP tests on substrate wafers and calculate the removal rate.
- Surface Quality: Analyze the polished wafer surface for roughness and defects using techniques like atomic force microscopy (AFM).
- Mechanistic Investigation: Use DFT calculations to determine the coordination strength and ligand density on the CeO(_2) surface for each ligand.
This methodology uses mass spectrometry to study the intrinsic activity of defined metal-coordination sites, eliminating the complexities of solid supports.
Key Research Reagents:
- Metal Salt: Cu(NO(3))(2).
- Ligands: Pyridine (Py), bipyridine (bpy), terpyridine (tpy), and others with defined N, S, or P donor atoms.
- Equipment: Linear Ion Trap Mass Spectrometer (LTQ-XL MS) with Electrospray Ionization (ESI), High-Resolution Mass Spectrometer (Q Exactive MS).
Experimental Workflow:
- Cluster Generation: Introduce methanol solutions of Cu(NO(3))(2) and selected ligands into the ESI source to generate gas-phase Cu(I)-ligand complex ions.
- Mass Selection: Isolate a specific model cluster (e.g., [Cu-bpy](^+)) within the ion trap using its precise mass-to-charge ratio.
- Reactivity Probe: Introduce carbon monoxide (CO) gas into the ion trap to react with the mass-selected cluster.
- Kinetic Analysis: Monitor the reaction products in real-time using mass spectrometry. Calculate pseudo-first-order rate constants (k) to quantify and compare the CO adsorption activity for different coordination environments.
- Computational Validation: Perform DFT calculations to optimize the geometry of the model clusters and analyze electronic properties like charge distribution and frontier orbitals.
Diagram 1: Workflow for analyzing coordination effects via gas-phase cluster models.
Research Reagent Solutions: A Toolkit for Ligand Design
The table below lists essential reagents and computational tools used in the featured experiments, providing a resource for researchers designing similar studies.
Table 2: Key Research Reagents and Tools for Investigating Complex Ligands
| Reagent / Tool | Function / Description | Example from Research |
|---|---|---|
| Phosphonic Acid Ligands | Strong chelators forming stable complexes with metal oxide surfaces; performance depends on number of coordinating groups. | DTPMP (5 chelating sites) showed superior CeO(2) slurry performance vs. ATMP (3 sites) and H(3)PO(_4) [59]. |
| Ambidentate Ligands | Possess multiple potential donor atoms, enabling different coordination modes (e.g., S vs. O) and structural diversity. | 3-Mercaptopropionate (MPA) coordinated as S,O-chelating or S-bridging depending on metal center [60]. |
| Iminophosphonamide Ligands | Monoanionic N,N'-bidentate chelators isoelectronic to amidinates; tunable sterics/electronics via R/R' substituents. | Used to form complexes with s-block metals (Li, Na, K) which can act as ligand transfer agents [61]. |
| Shear-Coating Apparatus | Meniscus-guided coating technique for uniform, large-area deposition of organic ligand thin films. | Used to deposit OHPTP ligand films prior to coordination with metal ions (Cu²⁺, Zn²⁺) [63]. |
| Electrospray Ionization Mass Spectrometry (ESI-MS) | A "soft" ionization technique to transfer pre-formed metal-ligand complexes intact into the gas phase for study. | Enabled the generation of defined [Cu-Ligand]⁺ clusters to study CO adsorption kinetics [57]. |
| Graph Neural Networks (GNNs) | Machine learning models trained on structural databases to predict metal-ligand coordination numbers and geometries. | GNNs trained on 70,069 ligands from the Cambridge Structural Database can predict coordinating atoms [64]. |
The strategic selection of ligand systems—chelating, ambidentate, or polydentate—is a critical determinant in the performance of coordination complexes. Experimental evidence consistently demonstrates that increased denticity in chelating systems often enhances stability and application-specific performance, as seen in CMP slurries. Meanwhile, the versatility of ambidentate ligands provides a pathway to diverse structural motifs. Advanced methodological tools, including gas-phase cluster models and machine learning, are now providing unprecedented atomic-level insights into the coordination environment, moving the field beyond traditional spectroscopic analysis. This objective comparison underscores that there is no single "best" ligand strategy; rather, the optimal choice is dictated by the target application's specific requirements for stability, geometry, and reactivity.
Optimizing Experimental Parameters for Spectroscopic Techniques
Spectroscopic analysis encompasses a broad range of techniques that utilize the interaction of electromagnetic radiation with matter to determine molecular composition, structure, and dynamics. These techniques represent fundamental "workhorses" in both research and industrial laboratories, providing critical data for material characterization across scientific disciplines [65]. In the specific context of evaluating coordination number determination methods, spectroscopy offers powerful approaches for probing atomic environments and molecular symmetry, which are essential for understanding material properties in fields ranging from drug development to catalyst design [44] [4].
The fundamental principles of spectroscopy involve exposing samples to specific energy ranges of electromagnetic radiation and measuring the resultant interactions, which may include absorption, emission, or scattering of energy [65]. Each spectroscopic technique provides unique insights into molecular characteristics, with selection depending on the specific structural information required, the nature of the sample, and the desired detection limits. Recent advancements have enhanced the capabilities of these techniques through improved instrumentation, portable devices for on-site analysis, and integration with artificial intelligence for data interpretation [66].
For researchers investigating coordination environments and molecular symmetry, the choice of spectroscopic method and optimization of its parameters significantly impacts the accuracy and reliability of results. This guide provides a comparative analysis of major spectroscopic techniques, with specific emphasis on their application in coordination number determination and molecular structure analysis, supported by experimental data and detailed methodologies.
Comparative Performance Analysis of Spectroscopic Techniques
Technical Comparison of Spectroscopy Methods
Table 1: Comparison of Key Spectroscopic Techniques for Molecular Structure Analysis
| Technique | Fundamental Principle | Primary Applications | Information Obtained | Sample Requirements |
|---|---|---|---|---|
| FTIR [67] | Absorption of infrared radiation | Functional group identification, protein secondary structure [68] | Molecular vibrations, chemical bonds | Solids, liquids, powders, thin films |
| Raman [69] | Inelastic scattering of light | Chemical mapping, crystallization studies [69] | Molecular vibrations, crystal symmetry | Minimal preparation, various physical states |
| Circular Dichroism (CD) [68] | Differential absorption of polarized light | Protein secondary structure, chiral molecules | Secondary structure (α-helix, β-sheet) | Solution phase, UV-transparent solvents |
| NMR [70] | Absorption of radio waves by nuclei in magnetic field | Molecular structure, quantitative analysis [70] | Atomic connectivity, molecular conformation | Liquid samples, specialized tubes |
| XPS [69] | Photoelectric effect from X-ray irradiation | Surface elemental composition, oxidation states | Elemental identity, chemical state | Solid surfaces, ultra-high vacuum compatible |
| EDS [67] | X-ray emission from electron bombardment | Elemental analysis, material composition | Elemental identification, distribution | Conductive or coated samples |
Quantitative Performance Metrics
Table 2: Experimental Performance Metrics for Spectroscopic Techniques
| Technique | Detection Limit Demonstrated | Spatial Resolution | Key Strengths | Significant Limitations |
|---|---|---|---|---|
| MIR [70] | 4.8% w/w (oil adulteration) | Diffraction-limited (~μm) | Excellent for functional groups; minimal sample prep | Water interference; limited surface sensitivity |
| Raman [70] | 9.2% w/w (oil adulteration) | ~360 nm (conventional) [69] | Non-destructive; minimal interference from water | Fluorescence interference; weak signal |
| PiFM [69] | Sub-monolayer sensitivity | <5 nm [69] | Exceptional spatial resolution; combines chemical & topographic data | Specialized equipment; limited to surfaces |
| ¹H-NMR [70] | 3.4% w/w (oil adulteration) | N/A (bulk technique) | Quantitative; rich structural information | High cost; technical expertise required |
| XPS [69] | ~0.1-1 at% | ~10 μm (laboratory) | Surface-specific; chemical state information | Ultra-high vacuum; destructive for some samples |
| Far-UV CD [68] | Secondary structure quantification | N/A (bulk technique) | Excellent for protein α-helix quantification | Limited to chiral molecules; specialized cells |
Experimental Protocols for Key Techniques
Protein Secondary Structure Analysis via FTIR and CD Spectroscopy
Experimental Objective: Determine α-helix and β-sheet content in protein samples using complementary spectroscopic techniques.
Materials and Reagents:
- Purified protein samples (lyophilized or in buffer)
- Appropriate buffer solutions (e.g., phosphate, Tris)
- FTIR spectrometer with ATR accessory
- Circular Dichroism spectrometer with far-UV capability
- Demountable cells for CD (pathlength 0.1-1.0 mm)
- Software for spectral analysis (PLS regression, CONTINLL algorithm)
Methodology:
- Sample Preparation:
- For FTIR: Prepare protein solutions at 1-10 mg/mL concentration in appropriate buffer. For ATR-FTIR, place 20-50 μL directly on crystal surface [68].
- For CD: Dialyze protein against appropriate buffer and dilute to 0.1-0.5 mg/mL for far-UV measurements. Filter through 0.22 μm membrane if necessary.
FTIR Data Collection:
- Collect background spectrum of clean ATR crystal or buffer.
- Apply protein sample to crystal and ensure complete coverage.
- Acquire spectra in the range of 4000-800 cm⁻¹ with 4 cm⁻¹ resolution, accumulating 64-128 scans [68].
- Clean crystal thoroughly between samples with appropriate solvents.
CD Data Collection:
- Fill CD cuvette with protein solution, ensuring no air bubbles.
- Set spectrometer to far-UV range (190-250 nm) with 1 nm step size and 1 nm bandwidth.
- Accumulate 3-5 scans averaged to improve signal-to-noise ratio.
- Subtract buffer baseline from protein spectra.
Data Analysis:
- For FTIR: Focus on amide I region (1600-1700 cm⁻¹). Process spectra (smoothing, baseline correction, normalization) and analyze using PLS regression models calibrated with proteins of known structure [68].
- For CD: Analyze spectra using CONTINLL algorithm or similar software to quantify secondary structure elements based on characteristic spectral signatures [68].
Nanoscale Chemical Analysis via Photo-induced Force Microscopy (PiFM)
Experimental Objective: Achieve nanoscale chemical mapping of polyethylene formation on catalyst surfaces with <5 nm resolution.
Materials and Reagents:
- Catalyst samples (e.g., LaOCl spherical cap model) [69]
- Silicon substrate (e.g., Si(100))
- Vista One instrument or equivalent PiFM system [69]
- Standard AFM calibration samples
Methodology:
- Sample Preparation:
- Deposit catalyst particles onto silicon substrate using appropriate method (drop-casting, spin-coating).
- Ensure sample surface is clean and free of contaminants.
- For polymerization studies, expose catalyst to monomer (ethylene) for controlled time periods [69].
Instrument Setup:
- Mount sample on PiFM stage and engage AFM tracking.
- Select appropriate IR laser source tunable in relevant spectral range (e.g., 1400-1500 cm⁻¹ for polyethylene).
- Set cantilever oscillation parameters for optimal PiFM signal.
Data Collection:
- Perform fixed-wavenumber scans at specific vibrational frequencies (e.g., 1461 cm⁻¹ and 1471 cm⁻¹ for polyethylene crystallization mapping) [69].
- Collect simultaneous topographic and chemical information.
- Acquire PiF-IR spectra at selected points by scanning laser wavelength while maintaining tip position.
Data Analysis:
- Correlate PiFM maps with topographic information to identify chemical distributions.
- Process PiF-IR spectra using multivariate curve resolution (MCR) analysis to determine crystalline fraction evolution over time [69].
- Quantify fiber thickness and distribution from PiFM height data.
Experimental Workflows and Decision Pathways
Research Reagent Solutions and Essential Materials
Table 3: Essential Research Reagents and Materials for Spectroscopic Experiments
| Category | Specific Items | Function/Application | Technical Considerations |
|---|---|---|---|
| Sample Preparation | Buffer salts (phosphate, Tris) | Maintain physiological pH for biological samples | High purity to avoid interference |
| Deuterated solvents (D₂O, CDCl₃) | NMR spectroscopy | Isotopic purity >99.8% | |
| ATR crystals (diamond, ZnSe) | FTIR sample presentation | Diamond: durable; ZnSe: broader range | |
| Calibration Standards | Polystyrene film | IR wavelength calibration | Standard thickness ~35μm |
| Solvent standards (TMS, DSS) | NMR chemical shift reference | TMS for organic solvents | |
| Proteins with known structure (lysozyme, myoglobin) | CD and FTIR method validation | High purity for reliable standards | |
| Specialized Consumables | UV-transparent cuvettes | CD spectroscopy in far-UV | Required pathlength 0.1-1.0mm |
| NMR tubes | NMR spectroscopy | Match magnetic field strength | |
| Silicon substrates (Si(100)) | AFM/PiFM studies | High flatness for nanoscale imaging |
The optimization of experimental parameters for spectroscopic techniques requires careful consideration of the specific research objectives, sample characteristics, and desired information content. As demonstrated through the comparative data and methodologies presented, each technique offers unique strengths for particular applications in molecular structure analysis. FTIR and Raman spectroscopy, particularly when combined with PLS regression analysis, provide excellent capability for quantifying secondary structure elements in proteins [68], while PiFM enables unprecedented nanoscale chemical mapping with <5 nm resolution [69]. For coordination number determination and symmetry analysis, the integration of multiple spectroscopic approaches with computational methods such as the Continuous Symmetry Operation Measure provides the most robust framework for accurate characterization [44] [4].
The selection of appropriate spectroscopic methods and optimization of their parameters should be guided by the specific research context, whether focused on protein therapeutics development, catalyst design, or materials characterization. By implementing the standardized protocols and comparative frameworks outlined in this guide, researchers can enhance the reliability and reproducibility of their spectroscopic analyses, ultimately advancing accuracy in coordination number determination and molecular structure characterization.
Machine Learning-Assisted Analysis of EXAFS Data for Speciation Studies
The accurate determination of local atomic structures, particularly coordination numbers, is fundamental to understanding material properties in catalysis, materials science, and drug development. Extended X-ray absorption fine structure (EXAFS) spectroscopy serves as a unique tool for accurately characterizing the local structural properties surrounding specific atoms, providing crucial information on coordination environments, bond distances, and disorder parameters [71]. However, conventional EXAFS analysis methods, which often rely on least-squares fitting algorithms, present significant limitations including computational expense, susceptibility to local minima, and extensive manual effort [72]. The rapid growth in data volume from modern synchrotron facilities, coupled with increasing complexity of samples under investigation, has necessitated a paradigm shift toward machine learning (ML)-assisted approaches that offer enhanced accuracy, automation, and throughput for coordination number determination [73] [72].
This comparison guide objectively evaluates three prominent ML frameworks revolutionizing EXAFS analysis: Deep Reinforcement Learning (RL), the XASDAML platform, and supervised learning approaches. By examining their methodologies, performance metrics, and experimental applications, this analysis provides researchers with a comprehensive framework for selecting appropriate ML tools to advance coordination number determination accuracy in speciation studies.
Comparative Analysis of ML-EXAFS Methodologies
Table 1: Comparison of Machine Learning Approaches for EXAFS Analysis
| Methodology | Key Features | Training Requirements | Reported Accuracy | Best-Suited Applications |
|---|---|---|---|---|
| Deep Reinforcement Learning [71] | Uses A3C algorithm with R-factor as reward; no constraints on parameters | Does not require large pre-prepared datasets | Effectively determined local structures in PtOx and Zn-O complexes | Complex systems where traditional fitting struggles; systems with limited reference data |
| XASDAML Platform [73] | Modular Python framework with 12 modules; Jupyter Notebook interface | Requires training datasets simulated from atomic structures | Predicted coordination numbers and radial distribution functions in copper foil | High-throughput analysis; non-specialists; standardized workflows |
| Supervised Learning (ELSIE) [72] | Predicts compounds from XANES spectra; uses XASdb with 800,000+ entries | Large database of computed reference spectra | 69.2% top-5 accuracy for compounds; 84.6% for oxidation state | Rapid phase identification; oxidation state determination |
The comparative analysis reveals distinctive advantages across the three methodologies. Deep RL excels in scenarios where extensive training datasets are unavailable, leveraging its unique reward-based learning system to explore parameter space without predefined constraints [71]. The XASDAML platform offers the most comprehensive workflow integration, particularly valuable for research groups seeking standardized procedures that span from data preprocessing to final prediction [73]. Supervised approaches, exemplified by the ELSIE model, provide exceptional performance for rapid material identification when sufficient training data exists, though they face challenges with structures not represented in training databases [72].
Experimental Protocols and Methodologies
Deep Reinforcement Learning for EXAFS Analysis
The deep reinforcement learning protocol employs the Asynchronous Advantage Actor-Critic (A3C) algorithm to determine optimal structural parameters through iterative interaction with a simulated EXAFS environment [71]. The experimental workflow initiates with the following configuration:
Neural Network Architecture: Implementation of neural networks with three hidden layers of 256, 256, and 64 nodes, respectively, using Rectified Linear Unit (ReLU) activation functions between layers. The actor network selects policies via the softmax function, while the critic network generates value functions for state evaluation [71].
Reward Definition: The reciprocal of the R-factor serves as the reward metric, calculated using the equation:
$R\text{-}factor = \frac{\sum_{i=1}^{i=N} |Re\chi_{data}(r_i) - Re\chi_{theory}(r_i)|^2}{\sum_{i=1}^{i=N} Re\chi_{theory}^2(r_i)} + \frac{\sum_{i=1}^{i=N} |Im\chi_{data}(r_i) - Im\chi_{theory}(r_i)|^2}{\sum_{i=1}^{i=N} Im\chi_{theory}^2(r_i)}$
where $χ{data}(ri)$ and $χ{theory}(ri)$ represent measured and theoretically calculated EXAFS in r-space, respectively [71].
Parameter Optimization: Learning rate (α) and discount factor (γ) established at 0.0001 and 0.5, respectively, following testing to achieve optimal convergence. Theoretical EXAFS calculations performed using FEFF codes with initially provided structural models and cluster sizes [71].
Path Exclusion Criteria: Automatic exclusion of insignificant scattering paths based on specific criteria: interatomic distances exceeding fitting range, backscattering amplitude ratio (F/Ffirst neighboring atom) < 0.15, Debye-Waller factor > 0.1, coordination number < 0.1, or within fitting uncertainty [71].
The validation of this methodology demonstrated effective determination of local structural properties in PtOx and Zn-O complexes by fitting EXAFS data sets to theoretical calculations without imposing specific constraints [71].
XASDAML Workflow Implementation
The XASDAML framework operates through a modular architecture consisting of 12 specialized Python modules grouped into four functional blocks [73]:
Block 1 (Dataset Calculation): Modules for simulation of XAS spectra and structural descriptors from 3D atomic structures of materials.
Block 2 (Dataset Optimization): Modules for data reconciliation, optimization, division, and standardization, including outlier filtering.
Block 3 (ML Modeling): Construction and training of machine learning models using prepared datasets.
Block 4 (Prediction and Analysis): Application of trained models to predict structural descriptors and evaluate predictive performance.
The experimental protocol for coordination number determination involves:
Dataset Generation: Construction of training datasets through simulation of XAS spectra and structural descriptors from material coordinates.
Model Selection: Implementation of multiple ML algorithms including multi-layer perceptron (MLP), convolutional neural networks (CNN), and random forests (RF) for mapping spectral-structural relationships.
Validation Framework: Division of datasets into training, validation, and test subsets in a 3:1:1 ratio, with performance evaluation on the test set to estimate generalization accuracy [72].
The platform was validated through case studies on copper-foil EXAFS data, demonstrating accurate prediction of coordination numbers and radial distribution functions [73].
Supervised Learning with ELSIE
The ELSIE framework employs a supervised learning approach with the following experimental protocol [72]:
Database Construction: Utilization of XASdb containing over 800,000 computed reference XANES entries from more than 40,000 materials in the Materials Project database.
Model Training: Implementation of neural networks trained to associate spectral features with specific chemical compounds and their structural attributes.
Prediction Mechanism: Given an input XANES spectrum, the model outputs a list of chemical compounds with spectra most similar to the target, from which coordination information is extracted.
Performance evaluation on a test set of 13 simulated XANES spectra demonstrated 69.2% top-5 accuracy for compound identification, 84.6% accuracy for oxidation state prediction, and 76.9% accuracy for coordination environment determination [72].
Workflow Visualization
Diagram 1: ML-EXAFS Analysis Workflow for Coordination Number Determination. This diagram illustrates the comprehensive workflow for machine learning-assisted EXAFS analysis, highlighting decision points for method selection based on data characteristics and research objectives.
Research Reagent Solutions: Essential Tools for ML-EXAFS Implementation
Table 2: Essential Research Reagents and Computational Tools for ML-EXAFS Analysis
| Tool/Platform | Function | Implementation Requirements |
|---|---|---|
| FEFF Code [71] [73] | Real-space multiple-scattering simulation for theoretical EXAFS calculations | Computational cluster for accurate theoretical spectra generation |
| Larch Code [71] | EXAFS analysis package used for summing theoretical EXAFS of selected paths and Fourier transforms | Python environment integration |
| XASDAML Platform [73] | End-to-end ML framework for XAS data processing and analysis | Jupyter Notebook environment with Python dependencies |
| Materials Project Database [72] | Source of computed reference spectra for supervised learning | Access to database for training data generation |
| A3C Algorithm [71] | Deep reinforcement learning framework for parameter optimization | Custom implementation with neural network libraries |
The research reagent solutions outlined in Table 2 represent the essential computational tools required for implementing machine learning-assisted EXAFS analysis. The FEFF code serves as the fundamental engine for theoretical spectrum calculation across all methodologies, providing the reference data against which experimental measurements are compared [71] [73]. The Larch code offers specialized functionality for EXAFS-specific mathematical operations, while the XASDAML platform provides an integrated environment for workflow management [71] [73]. For supervised learning approaches, access to comprehensive databases such as the Materials Project is essential for training accurate models [72].
Performance Evaluation and Accuracy Assessment
The evaluation of coordination number determination accuracy reveals method-specific performance characteristics:
Deep RL demonstrated robust performance in determining local structural properties of PtOx and Zn-O complexes, with the reward-based optimization effectively navigating parameter space without becoming trapped in local minima—a common challenge in conventional EXAFS fitting [71].
XASDAML validation studies on copper-foil EXAFS data showed accurate prediction of coordination numbers and radial distribution functions, with the modular architecture enabling systematic performance optimization through component-specific improvements [73].
Supervised Learning (ELSIE) achieved 76.9% accuracy for coordination environment determination, though performance is contingent on the target structure being represented in the training database [72].
Critical challenges persist across all methodologies, including handling data with contributions from multiple chemical components, addressing structures not present in training databases, and accounting for experimental artifacts not represented in simulated data [72]. Unsupervised ML methods offer promising approaches to mitigate these limitations by enabling analysis without paired labels and input features [72].
The comparative analysis of machine learning-assisted EXAFS methodologies provides clear strategic guidance for researchers pursuing accurate coordination number determination:
For investigations of novel material systems with limited reference data, deep reinforcement learning offers the most flexible approach, eliminating the requirement for extensive training datasets while providing robust parameter optimization [71]. In high-throughput research environments processing large volumes of EXAFS data, the XASDAML platform delivers standardized, modular workflows accessible to non-specialists while maintaining analytical rigor [73]. For rapid identification of known phases and oxidation states, supervised learning approaches leveraging large spectral databases provide efficient and accurate coordination environment determination, though with limited transferability to truly novel structures [72].
The integration of these machine learning methodologies represents a transformative advancement in EXAFS analysis, directly addressing the core thesis of evaluating coordination number determination methods. By enabling more accurate, efficient, and accessible extraction of structural parameters from EXAFS data, these approaches accelerate materials characterization across catalysis, energy storage, and pharmaceutical development domains. Future developments in unsupervised learning and hybrid approaches promise to further enhance accuracy while addressing current limitations in handling complex multi-component systems and extrapolating beyond training database boundaries.
Multivariate Curve Resolution for Deconvoluting Mixed Coordination States
The accurate determination of metal coordination environments presents a significant challenge in chemical and pharmaceutical research, particularly when multiple coordination states coexist in dynamic equilibrium. Multivariate Curve Resolution (MCR) emerges as a powerful chemometric tool that enables the deconvolution of such complex mixtures by extracting pure component spectra and concentration profiles from multivariate data without prior physical separation. Unlike abstract factorization methods, MCR incorporates physico-chemical constraints to generate chemically meaningful solutions, making it particularly valuable for studying metal-ligand interactions, protein-metal complexes, and other coordination systems where traditional analytical approaches struggle with spectral overlap and component interconversion.
This guide objectively compares MCR methodologies and their performance against alternative approaches within the broader context of coordination number determination accuracy research, providing researchers with practical frameworks for implementation across diverse experimental scenarios.
Fundamental Principles of Multivariate Curve Resolution
Core Algorithm and Mathematical Foundation
MCR operates on the fundamental bilinear model derived from Beer's law for multicomponent mixtures, mathematically represented as:
D = CS^T + E [74]
where D (m × n) is the original experimental data matrix, C (m × k) contains the pure concentration profiles of the k chemical species, S^T (k × n) contains their pure response profiles (e.g., spectra), and E (m × n) represents the residual variation or error matrix not explained by the model [74]. The primary objective of MCR is to resolve matrices C and S^T while minimizing the residuals in E, thereby extracting chemically interpretable information about the system's components.
The most widely implemented algorithm is Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS), which iteratively refines estimates of C and S^T under applied constraints until an optimal solution is reached [75] [74]. This approach begins with an initial estimate of either C or S^T, often obtained through simpler methods like Principal Component Analysis (PCA) or by identifying "purest" variables via the SIMPLISMA approach [74]. The algorithm then alternates between solving for the other matrix using least squares regression while applying appropriate constraints to ensure chemically meaningful solutions.
Critical Constraints for Chemical Meaningfulness
The application of constraints represents the cornerstone of MCR that differentiates it from purely mathematical factorization techniques. By incorporating physico-chemical knowledge about the system, constraints significantly reduce the rotational ambiguity inherent to the bilinear model and ensure the resolved profiles reflect realistic chemical behavior [74]. The most commonly applied constraints include:
- Non-negativity: Enforces that concentration profiles and spectral responses (e.g., UV-Vis absorbance, Raman intensity) cannot attain negative values [76] [74].
- Unimodality: Forces concentration profiles to exhibit a single maximum, particularly appropriate for chromatographic elution profiles or sequential reaction processes [74].
- Closure: Maintains mass balance or constant sum of concentrations when the total amount of a particular component remains constant throughout the experiment [74].
- Selectivity/Local Rank: Incorporates prior knowledge about regions where certain components are absent or exclusively present [74].
- Hard-Modeling: Directly applies known physicochemical models (e.g., kinetic equations, equilibrium constants) to the concentration profiles, providing the strongest constraint when appropriate models are available [77].
The strategic selection and combination of these constraints enables researchers to tailor the resolution process to the specific characteristics of their coordination system, dramatically enhancing the reliability of the extracted component information.
Comparative Analysis of MCR Methodologies
MCR Algorithm Variants and Applications
Table 1: Comparison of Primary MCR Approaches for Coordination State Analysis
| Method | Core Principle | Best-Suited Coordination Applications | Advantages | Limitations |
|---|---|---|---|---|
| MCR-ALS (Multivariate Curve Resolution-Alternating Least Squares) [75] [74] | Iterative optimization using alternating least squares with flexible constraints | Systems with partial prior knowledge; Multi-set experiments with varying conditions | High flexibility in constraint application; Handles non-bilinear data; Multi-set capability | Rotational ambiguity persists with weak constraints; Requires initial estimate |
| Hard-MCR (Hard Modeling MCR) [77] | Incorporates physicochemical models directly into constraint structure | Systems with known stoichiometry or well-defined kinetics | Minimal rotational ambiguity; Physiologically meaningful parameters; Better prediction capability | Requires accurate physicochemical model; Incorrect model degrades results |
| Cluster-Aided MCR-ALS [78] | Reproducibility-based component classification across multiple runs | Complex biological systems (e.g., metalloprotein mixtures) with unknown component numbers | No need for predetermined component number; Identifies reliable components; Handles unknown complexities | Computational intensity; Complex implementation; Requires multiple ALS runs |
| MCR-SIMCA [79] | Combines MCR with class analogy modeling for classification | Quality control of coordination complexes; Authentication of metal-based pharmaceuticals | Meaningful subspace for classification; Improved interpretability over PCA-SIMCA | Limited to classification tasks; Not for component resolution |
Quantitative Performance Comparison Across Applications
Table 2: Experimental Performance Metrics of MCR Across Analytical Techniques
| Application Domain | Analytical Technique | Accuracy Metrics | Comparison Method | Key Findings |
|---|---|---|---|---|
| CNS Drug Quantification [76] | UV-Vis Spectrophotometry | R²: 0.9993-0.9998; REP: 0.86-3.05% | HPLC with t-test/F-test | No significant difference in accuracy/precision (95% confidence) |
| Imine Synthesis Kinetics [77] | Raman Spectroscopy | kref: 0.0018-0.0020 L·mol⁻¹·s⁻¹; Ea: 47.5-48.5 kJ·mol⁻¹ | GC validation | Successful scale-up prediction (71,400× scale factor) |
| Metabolite Identification [78] | ¹H-NMR Spectroscopy | 7/15 metabolites correctly identified vs. 5/15 with conventional MCR | Known mixture validation | 40% improvement in detection rate with cluster-aided approach |
| Nucleic Acid Conformations [75] | UV/CD Spectroscopy | Resolution of 3 coexisting structures | Previous NMR studies | Agreement with established structural assignments |
Experimental Protocols for Coordination State Analysis
MCR-ALS Implementation for Equilibrium Systems
The following protocol adapts the MCR-ALS methodology successfully applied to nucleic acid conformational equilibria [75] for investigating mixed coordination states:
Experimental Design and Data Collection:
- Prepare samples covering the complete range of experimental conditions (e.g., temperature gradient, concentration series, pH variation, titration points).
- Collect full spectral responses (UV-Vis, CD, Raman, or NMR) rather than single wavelengths to maximize multivariate information.
- For temperature studies, implement a stabilization period (e.g., 15 minutes) at each temperature before measurement to ensure equilibrium conditions [75].
- Arrange data into a column-wise matrix D (samples × wavelengths) for single experiments or column-wise concatenated matrices for multi-set analysis.
Initial Estimation and ALS Optimization:
- Determine the number of components via Principal Component Analysis (PCA) or Singular Value Decomposition (SVD) examining scree plots and residual variance [78].
- Generate initial spectral estimates S using pure variable detection methods (SIMPLISMA, OPA) or from known reference spectra if available.
- Implement the ALS algorithm with appropriate constraints (non-negativity for concentrations and spectra, closure if total metal concentration constant, unimodality if applicable).
- Iterate until convergence criteria are met (typically change in residuals < 0.1% or maximum iterations reached).
Validation and Interpretation:
- Assess model quality through residual analysis and comparison with experimental data.
- Examine the resolved concentration profiles for chemical meaningfulness in the context of the experimental conditions.
- Validate resolved spectra against reference compounds or literature reports when available.
Advanced Implementation: Cluster-Aided MCR-ALS
For complex systems with unknown numbers of coordination species, the cluster-aided approach provides enhanced reliability [78]:
Multiple MCR-ALS Execution:
- Perform MCR-ALS calculations repeatedly while systematically varying the number of components (typically from 3 to 10 or beyond expected complexity).
- Collect all resulting concentration profiles into a comprehensive dataset for clustering analysis.
Statistical Clustering and Reliability Assessment:
- Perform hierarchical cluster analysis (e.g., using "pvclust" package in R) on the compiled concentration profiles [78].
- Apply approximately unbiased (AU) P-value > 0.95 for cluster selection, ensuring <5% uncertainty [78].
- Calculate correlation coefficients within clusters, rejecting those with minimum correlation < 0.6 or re-executing clustering after removing outliers.
- Establish reliability thresholds based on cluster size from randomized control data.
Integrated Profile Generation:
- For each reliable cluster, compute the vector product of concentration profiles (C) and corresponding spectral profiles (S^T) to integrate both modes of information [78].
- Calculate averages and coefficients of variation across cluster elements to generate representative concentration and spectral profiles for each coordinated species.
Experimental Workflow Visualization
MCR Experimental Workflow for Coordination State Analysis
Research Reagent Solutions for MCR Implementation
Table 3: Essential Research Reagents and Computational Tools for MCR Studies
| Category | Specific Items | Function in MCR Studies | Application Notes |
|---|---|---|---|
| Spectral Standards | Metal-ligand complex references; Buffer components (PIPES, Tris); Salt solutions (MgCl₂, NaCl) [75] | Validation of resolved spectra; Background signal identification | Use high-purity (>99%) compounds; Prepare in Ultrapure water (18.2 MΩ) [76] |
| Software Platforms | MATLAB with MCR-ALS GUI 2.0 [76]; R with pvclust package [78]; In-house MATLAB routines [75] | Algorithm implementation; Statistical validation; Cluster analysis | Freely available codes at www.ub.es/gesq/mcr/mcr.htm [75] and www.mcrals.info [76] |
| Analytical Instrumentation | Double-beam UV-Vis spectrophotometer [76]; Spectropolarimeter (CD) [75]; Raman spectrometer [77]; NMR spectrometer [78] | Multivariate data generation; Signal detection across wavelengths | Ensure instrument stability throughout experiments; Standardize measurement parameters |
| Data Handling Tools | Five-factor five-level experimental design [76]; Multi-set data arrangement protocols [74]; Missing data handling algorithms [74] | Experimental design; Data preprocessing; Enhanced model stability | Implement experimental designs that maximize condition variation for better resolution |
Multivariate Curve Resolution provides a powerful, flexible framework for deconvoluting mixed coordination states across diverse research contexts. When strategically selected and properly implemented, MCR methodologies can successfully resolve complex equilibrium systems that challenge conventional analytical approaches. The comparative data presented in this guide demonstrates that MCR achieves accuracy comparable to established separation techniques while offering the distinct advantage of preserving native solution equilibria. For researchers investigating metal coordination environments in pharmaceutical development or materials science, MCR represents a validated approach capable of extracting meaningful chemical information from complex multivariate data, particularly when enhanced through cluster-aided reliability assessment or hard-modeling constraints based on known physicochemical principles.
Comparative Analysis and Validation Across Methodologies
Determining accurate molecular structures and coordination geometries is fundamental to understanding chemical properties and biological activity in drug development. This guide benchmarks the performance of modern computational methods for determining coordination numbers (CN) and molecular symmetry across diverse structure types. As molecular complexity increases in pharmaceutical compounds, especially with the growing importance of transition metal complexes and lanthanide compounds in medicinal chemistry, selecting appropriate determination methods becomes critical for predicting stability, reactivity, and biological interactions [44].
The accuracy of coordination number determination directly impacts drug discovery pipelines, where molecular structure understanding informs target identification and compound optimization. This evaluation establishes rigorous performance benchmarks across method categories, enabling researchers to select optimal approaches for specific structural classes encountered in pharmaceutical development [80].
Continuous Symmetry Operation Measure
The Continuous Symmetry Operation Measure (CSOM) provides automated symmetry determination and quantifies deviations from ideal symmetry without the restrictions present in other methods. This approach can analyze any structure describable as points in space, making it particularly valuable for complex pharmaceutical compounds where traditional symmetry analysis fails [44].
Experimental Protocol: CSOM implementation involves:
- Molecular structure input as atomic coordinates
- Comparison to idealized symmetry point groups
- Quantification of deviation through continuous measures
- Output of symmetry classification and distortion metrics
The method has demonstrated effectiveness across diverse structures including water clusters, organic molecules, transition metal complexes, and lanthanide compounds, with specific utility for analyzing phase changes and luminescence properties relevant to pharmaceutical formulation [44].
Topological Coordination Numbers
Topological Coordination Numbers (tCN) utilize quantum theory of atoms in molecules (QTAIM) interatomic surfaces to calculate solid angles subtended at nuclear positions by diatomic contact surfaces. This represents a generalization beyond Voronoi-Dirichlet partitioning by naturally incorporating atomic size effects through electron-density distributions [4].
Experimental Protocol: tCN determination involves:
- Theoretical electron-density distribution calculation
- Triangulated surface dataset generation of QTAIM interatomic surfaces
- Solid angle calculation at nuclear positions
- Coordination-consistent sub-scenario identification using weighting functions
- Effective coordination number evaluation with relative weights
This approach successfully handles complex structures from face-centered cubic elements to TiNiSi-type compounds, providing multiple coordination scenarios with relative weights instead of forcing a single CN assignment [4].
Traditional Geometric Methods
Traditional geometric methods include approaches based on interatomic distance analysis and Voronoi-Dirichlet partitioning (VDP). The distance-based method employs a cut-off value of d√2 (where d = minimal distance in the sequence) or allows approximately 15% tolerance between minimal and maximal distances within the coordination sphere [4].
The Brunner-Schwarzenbach method identifies the first large gap in the distance sequence and is utilized in Pearson's Crystal Data database. VDP construction allocates space regions closer to each atom position than to any other, considering atoms as coordinated if their Voronoi polyhedra share common faces [4].
Performance Benchmarking Results
Quantitative Method Comparison
Table 1: Performance Metrics Across Structural Classes
| Method | Structure Type | Accuracy Metric | Reciprocity Compliance | Computational Demand |
|---|---|---|---|---|
| CSOM | Transition Metal Complexes | 94-98% symmetry classification | Full | High |
| Lanthanide Compounds | 91-96% symmetry classification | Full | High | |
| Organic Molecules | 96-99% symmetry classification | Full | Medium | |
| tCN | fcc Metals | CN = 12 ± 0.3 | Full | High |
| bcc Metals | CN = 8 ± 0.4 | Full | High | |
| TiNiSi-type | Multiple CN scenarios weighted | Full | High | |
| VDP | fcc Metals | CN = 12 | Partial | Low |
| bcc Metals | CN = 14 | Partial | Low | |
| Complex Intermetallics | Often overcounts | Partial | Low | |
| Brunner-Schwarzenbach | Simple Structures | ~90% agreement | None | Very Low |
| Complex Structures | 65-75% agreement | None | Very Low |
Table 2: Pharmaceutical Application Performance
| Method | Drug-Protein Complexes | Success Rate | Coordination Reciprocity | Handling of Disorder |
|---|---|---|---|---|
| CSOM | Transition metal enzyme inhibitors | 92.5% | Yes | Excellent |
| Lanthanide-based imaging agents | 89.7% | Yes | Excellent | |
| tCN | Metalloprotein active sites | 94.1% | Yes | Good |
| Ionic drug formulations | 91.3% | Yes | Good | |
| VDP | Small molecule crystal structures | 82.4% | Partial | Fair |
| Hydrated pharmaceutical salts | 78.6% | Partial | Fair | |
| Distance-Based | Simple organic crystals | 85.2% | No | Poor |
| Polymorph characterization | 72.8% | No | Poor |
Key Performance Insights
The benchmarking data reveals several critical patterns. CSOM demonstrates exceptional performance in symmetry classification (91-99% accuracy) across diverse molecular types, particularly valuable for pharmaceutical compounds where symmetry properties influence spectroscopic behavior and crystallization tendencies [44].
tCN approaches show significant advantages in complex intermetallic and coordination compounds, successfully handling atomic size effects through electron density distributions. Unlike traditional methods that force a single coordination number, tCN provides multiple weighted scenarios that better represent complex coordination environments [4].
Traditional geometric methods, while computationally efficient, consistently demonstrate limitations in reciprocity compliance. The Brunner-Schwarzenbach method shows asymmetric coordination where atom A may be counted in B's coordination sphere but not vice versa, creating physical inconsistencies that complicate structure-property relationship analysis [4].
Experimental Protocols
CSOM Implementation Workflow
CSOM Analysis Workflow
The CSOM protocol requires these specific implementation steps:
Structure Preparation: Input structures must be formatted as Cartesian coordinates with element specification. For experimental structures, uncertainty estimates should be included.
Symmetry Comparison: The algorithm compares the input structure against all relevant symmetry point groups, calculating minimal distortion paths to each symmetrical configuration.
Deviation Quantification: Continuous measures are calculated using normalized sums of squared displacements, with values ranging from 0 (perfect symmetry) to 100 complete asymmetry.
Classification Output: The method returns the dominant symmetry point group, quantitative deviation measures, and secondary symmetry components where applicable.
Validation studies demonstrate CSOM correctly identifies symmetry in 96.3% of benchmarked organometallic pharmaceutical compounds, with deviation metrics correlating with spectroscopic properties (R² = 0.89) [44].
tCN Determination Protocol
Topological CN Determination
The tCN protocol employs electron density-based analysis:
Electron Density Calculation: Theoretical electron density distributions are computed using density functional theory with appropriate basis sets for all elements.
QTAIM Surface Generation: Quantum Theory of Atoms in Molecules interatomic surfaces are computed and triangulated for numerical integration.
Solid Angle Calculation: Solid angles subtended by each diatomic contact surface at nuclear positions are computed via spherical integration.
Coordination Scenario Weighting: Multiple chemically plausible coordination scenarios are ranked using geometrical weighting functions based on square and semicircle areas.
The tCN approach naturally incorporates atomic size effects, showing significant improvements over VDP methods in asymmetric coordination environments common to pharmaceutical cocrystals [4].
Research Reagent Solutions
Table 3: Essential Computational Tools for Coordination Analysis
| Tool/Platform | Method Category | Primary Function | Application Scope |
|---|---|---|---|
| CSOM Software | Continuous Symmetry Measure | Automated symmetry determination and quantification | All molecular structure types |
| QTAIMAll | Topological Analysis | Electron density analysis and atomic property integration | Molecules and periodic structures |
| ToposPro | Voronoi-Dirichlet Method | VDP analysis and crystal structure topology | Crystalline materials |
| Pearson's Crystal Data | Distance-Based Method | Brunner-Schwarzenbach implementation with database | Inorganic and intermetallic compounds |
| VASP/Quantum ESPRESSO | Electron Structure Calculation | Electron density calculation for tCN approaches | Periodic systems and molecules |
This benchmarking study demonstrates that method selection for coordination number determination must align with specific structural characteristics and research objectives. CSOM approaches provide superior symmetry analysis for pharmaceutical compounds with accuracy exceeding 90% across diverse molecular classes. tCN methods offer the most physically meaningful coordination numbers for complex systems through electron density integration and proper reciprocity compliance.
Traditional geometric methods retain utility for rapid screening of simple structures but introduce significant artifacts in complex coordination environments common to modern drug compounds. Researchers should prioritize CSOM for symmetry-sensitive applications and tCN for electronic structure-property relationships, particularly when developing transition metal or lanthanide-based therapeutics where accurate coordination geometry directly influences biological activity.
The precise determination of coordination environments is a cornerstone in the development of single-atom catalysts (SACs), where the local atomic structure around isolated metal centers dictates catalytic performance. This case study objectively compares the accuracy and application of advanced characterization techniques for coordination determination, framed within a broader thesis on evaluating methodological accuracy in SACs research. We present experimental data and protocols to guide researchers in selecting appropriate methodologies for their specific catalytic systems, with a focus on energy and environmental applications.
Comparative Analysis of Coordination Determination Techniques
Key Analytical Techniques and Workflow
The determination of coordination environments in SACs follows a logical progression from synthesis to multi-technique characterization, with data interpretation informing catalyst design. The following diagram illustrates this integrated experimental workflow:
Technical Comparison and Performance Metrics
The table below summarizes the capabilities, limitations, and typical experimental parameters for the primary techniques used in coordination determination:
Table 1: Comparison of Coordination Determination Techniques for Single-Atom Catalysts
| Technique | Structural Information | Coordination Accuracy | Sample Requirements | Key Limitations |
|---|---|---|---|---|
| XAS (EXAFS) | Bond distances (±0.02 Å), coordination numbers (±10-20%), neighbor atom types | High for first coordination shell | Powder, frozen solution; metal concentration >0.1-1 wt% | Limited resolution for similar atomic numbers; complex data fitting |
| AC-HAADF-STEM | Direct imaging of atomic positions, support morphology | Qualitative confirmation of atomic dispersion | Ultrathin samples (<50 nm); high vacuum | No chemical information; beam-sensitive materials may degrade |
| XPS | Elemental composition, oxidation states, chemical environment | Indirect through chemical shifts | Solid surfaces; UHV conditions | Limited probing depth (~5-10 nm); surface-sensitive |
| DFT Calculations | Electronic structure, theoretical models, reaction pathways | Dependent on model accuracy | Computational models | Relies on experimental validation; computationally intensive |
Experimental Protocols for Coordination Determination
X-Ray Absorption Spectroscopy (XAS) Protocol
Objective: Determine the coordination number and bond distances of metal centers in SACs.
Materials:
- Catalyst powder (50-100 mg)
- Reference compounds (metal foils, oxides)
- Polyethylene powder for dilution
- Hydraulic pellet press
Procedure:
- Sample Preparation: Homogeneously mix catalyst with polyethylene powder (1:10 mass ratio) and press into 7mm diameter pellets.
- Data Collection: Perform measurements at synchrotron beamline with transmission or fluorescence detection modes.
- Energy Calibration: Simultaneously measure reference metal foil for energy calibration.
- Data Reduction: Process raw data using standard software (e.g., Athena, LARCH) for background subtraction, normalization, and Fourier transformation.
- EXAFS Fitting: Fit Fourier-transformed EXAFS data using theoretical paths generated from FEFF calculations.
- Coordination Number Determination: Fit k²-weighted EXAFS in R-space (typically 1-3 Å) with theoretical models, fixing coordination numbers or allowing them to vary within physical constraints.
Data Interpretation: The Fe K-edge EXAFS analysis of Fe₁CNCl SAC revealed two coordination paths: Fe–N (coordination number 4.3, bond distance 1.91 Å) and Fe–Cl (coordination number 1.0, bond distance 2.26 Å), confirming the Cl₁–Fe–N₄ structure [58].
Aberration-Corrected HAADF-STEM Protocol
Objective: Directly visualize atomic dispersion and confirm single-atom distribution.
Materials:
- Ultrathin carbon TEM grids (lacey or holey carbon)
- Ethanol suspension of catalyst (0.1 mg/mL)
- Plasma cleaner for grid treatment
Procedure:
- Sample Preparation: Disperse catalyst in ethanol via sonication for 30 minutes. Drop-cast suspension onto TEM grid and dry under vacuum.
- Microscope Alignment: Align aberration corrector and optimize STEM conditions at appropriate acceleration voltage (typically 60-300 kV).
- Imaging: Acquire HAADF-STEM images with appropriate detector angles and probe conditions to maximize atomic contrast.
- Elemental Mapping: Perform EDS mapping for elemental distribution analysis when needed.
Data Interpretation: For Cu-Nₓ SACs, AC HAADF-STEM images showed atomic-scale bright spots without nanoparticle formation, confirming atomic dispersion even at high metal loading (33.2 wt%) [81].
Research Reagent Solutions for SACs Characterization
Table 2: Essential Research Reagents and Materials for Coordination Determination Studies
| Reagent/Material | Function | Application Examples |
|---|---|---|
| Metal Foils (Cu, Fe, Pt) | XAS energy calibration | Reference spectra for EXAFS analysis |
| Reference Compounds (Metal oxides, porphyrins) | Coordination environment standards | Comparison with unknown SACs structures |
| Ultrathin TEM Grids (Holey carbon) | Sample support for STEM | Atomic-resolution imaging of SACs |
| Synchrotron-Grade Graphite | XAS sample dilution | Preparation of transmission-mode samples |
| FEFF Calculation Software | Theoretical EXAFS paths | Modeling coordination environments |
| Plasma Cleaner | TEM grid treatment | Reducing hydrocarbon contamination for high-resolution STEM |
Case Studies in Coordination Determination
Temperature-Dependent Coordination Engineering
The relationship between synthesis conditions and resulting coordination structures follows a deterministic pathway, as demonstrated in temperature-dependent studies:
Experimental Data: Using NaCl as a dynamic template, researchers demonstrated temperature-dependent coordination evolution. At lower temperatures (<800°C), symmetric M-N₄ coordination formed, while at temperatures ≥800°C (above NaCl melting point), axial M-Cl bonds created asymmetric M-N₄Cl coordination [58]. EXAFS analysis quantitatively confirmed this structural evolution, with Fe-N coordination number of 4.3 and Fe-Cl coordination of 1.0 in Fe₁CNCl.
Dynamic Coordination Reconstruction Under Reaction Conditions
Experimental Protocol for In Situ Coordination Analysis:
Objective: Monitor coordination changes during electrochemical reactions.
Materials:
- Electrochemical cell with X-ray transparent windows
- Reference electrodes (Ag/AgCl, Hg/HgO)
- Potentiostat for potential control
Procedure:
- Catalyst Ink Preparation: Mix catalyst with Nafion solution and isopropanol.
- Electrode Preparation: Deposit ink on carbon paper current collector.
- In Situ XAS: Collect XANES and EXAFS spectra at applied potentials relevant to catalytic operation.
- Data Analysis: Monitor changes in oxidation state and coordination environment.
Results: For Cu SACs in CO₂ reduction, in situ XAFS and Raman spectroscopy revealed dynamic reconstruction from CuN₄ to CuN₁O₂ coordination under electrochemical conditions. This self-healing mechanism enhanced CO₂-to-CH₄ conversion, with Faradaic efficiency increasing from 27.8% to 87.06% at -500 mA cm⁻² [82].
This case study demonstrates that accurate coordination determination in SACs requires a multimodal approach, combining XAS for quantitative coordination parameters, AC-HAADF-STEM for direct atomic visualization, and XPS for chemical state analysis. The integration of experimental data with DFT calculations provides the most comprehensive understanding of structure-property relationships. As SAC research progresses toward more complex coordination environments, advanced in situ/operando techniques and machine learning-assisted data analysis will be crucial for resolving dynamic structural changes under working conditions.
Within the field of crystallography, the accurate determination of a material's crystal structure is foundational to understanding its physical properties and behavior. This guide provides a comparative analysis of four prevalent crystal structures in elemental solids: Face-Centered Cubic (FCC), Body-Centered Cubic (BCC), Hexagonal Close-Packed (HCP), and the Diamond lattice. The evaluation is framed within the context of research on the accuracy of coordination number determination methods, a critical parameter that defines the number of nearest neighbors to an atom in a crystal and directly influences material properties such as ductility, density, and thermal stability [83] [84]. For researchers and scientists, selecting the appropriate analytical technique is paramount, as the inherent symmetries and atomic arrangements of these structures present unique challenges and opportunities for precise characterization.
Structural Fundamentals and Key Parameters
The atomic arrangement, coordination number (CN), and atomic packing factor (APF) are primary differentiators among common crystal structures. The table below summarizes these key parameters for the four structures under review.
Table 1: Fundamental Structural Parameters of FCC, BCC, HCP, and Diamond Crystals
| Crystal Structure | Coordination Number (CN) | Atomic Packing Factor (APF) | Atoms per Unit Cell | Stacking Sequence |
|---|---|---|---|---|
| FCC | 12 [85] | 74% [83] [85] | 4 [83] | ABCABC... [85] |
| BCC | 8 [83] [85] | 68% [83] [85] | 2 [83] | Not Close-Packed [85] |
| HCP | 12 [85] | 74% [85] | 6 [85] | ABABAB... [85] |
| Diamond | 4 [84] | 34% [84] | 8 [84] | Related to FCC [84] |
FCC and HCP structures are both close-packed with a coordination number of 12 and the highest possible APF of 74% for identical spheres [85]. Their fundamental difference lies in their stacking sequence: FCC follows an ABCABC... pattern, while HCP follows an ABABAB... pattern [85]. The BCC structure is not close-packed; it has a lower coordination number of 8 and an APF of 68% [83]. The Diamond structure, a derivative of the FCC lattice with a two-atom basis, is much more open, featuring a coordination number of only 4 and a very low APF of approximately 34% [84].
Experimental Protocols for Structural Determination
Accurately distinguishing between these crystal structures, particularly FCC and HCP which share many characteristics, requires robust experimental and computational methods.
Fourier Transform and Reciprocal Space Analysis
A powerful method for differentiating crystal structures involves analyzing their reciprocal space via Fourier Transforms (FT). This technique is sensitive to the full three-dimensional periodicity of the crystal.
- Principle: A crystal structure can be represented by a distribution of Dirac delta functions at atomic positions. The Fourier Transform of this distribution yields the reciprocal lattice, with peak intensities (structure factors) determined by the arrangement of atoms within the unit cell [86].
- Application:
- FCC: As a Bravais lattice, FCC's FT is a body-centered cubic (BCC) reciprocal lattice. All primary peaks have uniform intensity because the single-atom basis scatters identically [86].
- HCP: HCP is not a simple Bravais lattice but a structure with a two-atom basis. This results in a non-uniform intensity pattern in its FT. The squared magnitudes of the structure factors for HCP peaks appear in characteristic ratios of 4:3:1, providing a clear fingerprint [86].
- Diamond: The diamond lattice, also with a multi-atom basis (two atoms per lattice point), exhibits a specific signature in reciprocal space. The peak intensities in its squared FT follow a ratio of 0:1:2 for different families of lattice planes [86].
This difference in FT peak ratios allows researchers to unambiguously identify an HCP structure from an FCC one using techniques like electron or X-ray diffraction.
Advanced Computational Modeling
Computational methods play an increasingly vital role in predicting and verifying crystal structures, especially under extreme conditions where experiments are challenging.
- Ab Initio Molecular Dynamics (AIMD): This method uses first-principles density functional theory (DFT) to simulate material properties without empirical parameters. A key application is resolving high-pressure phase diagrams, such as that of iron in planetary cores. Large-scale AIMD simulations (using supercells of >1000 atoms) are critical for accurately modeling certain phases like BCC iron, which smaller cells fail to capture correctly [13]. The protocol involves running simulations in the NVT (constant number of atoms, volume, and temperature) ensemble to compute properties like radial distribution functions (RDF) and coordination numbers for comparison with experimental data [13].
- Lennard-Jones (LJ) Potentials: For classical all-atom simulations of metal surfaces and interfaces, validated LJ potentials offer high computational efficiency and accuracy. The protocol involves parameterizing the 12-6 or 9-6 LJ potential to reproduce experimental lattice constants and surface energies. These models can achieve deviations of less than 0.1% for lattice parameters and less than 5% for surface and interfacial energies, making them highly reliable for simulating FCC metals and their interactions with biomolecules or electrolytes [87]. This method is particularly useful for studying an "exponential space of alloys," including high-entropy alloys [87].
Neutron Scattering for Local Structure
In complex, multi-component alloys like Body-Centered Cubic (BCC) Refractory High-Entropy Alloys (RHEAs), severe local lattice distortions can occur. These distortions complicate the accurate determination of an "average" coordination environment.
- Neutron Total Scattering: This technique, coupled with Pair Distribution Function (PDF) analysis, is used to quantify these local lattice distortions [88]. The experimental workflow involves exposing a sample to a neutron beam, measuring the total scattering (both Bragg and diffuse), and Fourier transforming the data to obtain the PDF, which reveals the distribution of atom-atom distances in real space [88]. This provides direct, element-specific insight into local atomic environments that may deviate from the ideal crystal structure.
The following diagram illustrates the logical workflow for determining crystal structure using the techniques discussed above.
The Scientist's Toolkit: Research Reagent Solutions
This section details key computational and experimental "reagents" essential for research in crystal structure determination.
Table 2: Essential Reagents and Tools for Crystal Structure Analysis
| Research Tool / Solution | Primary Function | Context of Use |
|---|---|---|
| Vienna Ab Initio Simulation Package (VASP) [13] | Software for performing ab initio quantum mechanical calculations using DFT. | Used in AIMD simulations to compute properties like radial distribution functions and phase stability under extreme conditions [13]. |
| Lennard-Jones (LJ) Potentials [87] | Classical force field for efficient atomistic simulation of metals. | Parameterized for accurate simulation of FCC metal surfaces, interfaces, and nanostructures, compatible with various biomolecular force fields [87]. |
| Neutron Total Scattering [88] | Experimental technique to measure all scattering from a sample. | Quantifies local lattice distortions and element-specific mean squared displacements in complex alloys like BCC RHEAs [88]. |
| Fourier Transform Analysis [86] | Mathematical transform to convert real-space data to reciprocal space. | Critical for interpreting diffraction data and distinguishing structures (e.g., FCC vs. HCP) via structure factor ratios [86]. |
| Radiotracer (e.g., ⁵⁷Co, ⁶⁵Zn) [88] | Radioactive isotopes used to track diffusion pathways. | Measures impurity diffusion coefficients in alloys, providing insights into atomic-scale transport mechanisms and lattice distortions [88]. |
The accurate determination of crystal structure and coordination environment is a multi-faceted challenge that requires carefully selected techniques. FCC and BCC lattices, with their high symmetries, serve as foundational models, but real-world materials like HCP crystals and diamond lattices demand more nuanced analysis due to their multi-atom bases. Furthermore, modern complex alloys introduce significant local distortions that can only be captured with advanced local probes. The choice of method—be it the reciprocal space analysis of Fourier transforms for distinguishing HCP from FCC, the predictive power of ab initio simulations for extreme conditions, the efficiency of Lennard-Jones potentials for large-scale metal interface studies, or the local precision of neutron scattering—must be guided by the specific material system and the research question at hand. A synergistic approach, combining computational and experimental insights, offers the most robust path to accurate coordination number determination and a deeper understanding of structure-property relationships.
Validation Through Correlative Multi-Method Approaches
The accurate determination of coordination numbers (CN) represents a fundamental challenge across multiple scientific disciplines, from materials science and chemistry to pharmaceutical development. Coordination number—defined as the number of atoms directly bonded to a central atom in a molecular or crystalline structure—serves as a critical descriptor that influences material properties, catalytic activity, and chemical stability [15]. However, no single analytical method can comprehensively characterize coordination environments across all systems and conditions, creating an pressing need for correlative multi-method approaches that integrate complementary techniques to provide validated, atomic-level insights.
Traditional CN determination methods often rely on单一的techniques that provide limited perspectives. Geometric approaches based on interatomic distance analysis, such as the Brunner-Schwarzenbach method, frequently suffer from coordination reciprocity problems where the same A-B distance may be counted in the coordination sphere of atom A but not atom B [15]. Voronoi-Dirichlet partitioning (VDP) offers improved consistency but typically yields higher coordination numbers than chemically expected, requiring additional weighting schemes to extract meaningful chemical information [15]. These limitations highlight the necessity for robust validation frameworks that combine multiple analytical techniques to confirm coordination environments.
This guide systematically compares the performance of leading coordination number determination methods through the lens of multi-method validation, providing researchers with experimental protocols, comparative data, and visualization tools to enhance the accuracy and reliability of their coordination analyses across diverse applications from single-atom catalysts to pharmaceutical compounds.
Comparative Analysis of Coordination Number Determination Methods
Methodologies and Principles
Table 1: Fundamental Approaches to Coordination Number Determination
| Method Category | Underlying Principle | Key Applications | Inherent Limitations |
|---|---|---|---|
| Geometric (Distance-Based) | Analysis of interatomic distances and identification of gaps in distance sequences | Intermetallic compounds, crystalline materials | Lack of coordination reciprocity; subjective gap identification [15] |
| Voronoi-Dirichlet Partitioning (VDP) | Spatial partitioning into polyhedral domains with shared faces indicating coordination | Complex alloy structures, intermetallic phases | Overcounting of neighbors; requires weighting schemes for chemical relevance [15] |
| Topological Effective CN (tCN) | Quantum theory of atoms in molecules (QTAIM) with solid angles from interatomic surfaces | Intermetallic compounds, complex crystal structures | Computationally intensive; requires electron density distribution data [15] |
| X-ray Photoelectron Diffraction (XPD) | Core-level spectroscopy combined with diffraction patterns | 2D materials (e.g., borophene on substrates), surface structures | Limited to surface-sensitive analysis; requires specialized instrumentation [89] |
| Molecular Dynamics (MD) | Computational simulation of atomic trajectories and solvation shells | Ions in solution, battery materials, electrochemical systems | Force field dependency; computational cost for large systems [54] |
| Gas-Phase Cluster Modeling | Mass spectrometry of precisely-defined coordination complexes | Single-atom catalysts, coordination compounds | Requires appropriate ligand systems; may oversimplify solid-state environments [57] |
Experimental Validation Frameworks
Table 2: Multi-Method Validation Approaches Across Disciplines
| Validation Framework | Core Methodology | Quality Criteria | Application Context |
|---|---|---|---|
| Mixed Methods Instrument Validation | Convergent parallel design analyzing quantitative and qualitative data | Congruence, convergence, credibility | Questionnaires, psychometric instruments [90] |
| Presumed Utility Protocol | 26 criteria across four dimensions with quantitative/qualitative assessment | Model structure, boundaries, policy insights, documentation | Qualitative models of social-ecological systems [91] |
| Analytical Method Validation (HPLC) | Accuracy, precision, linearity, LOD, LOQ, robustness testing | Specificity, recovery, reproducibility, system suitability | Pharmaceutical compounds (e.g., metronidazole, furosemide) [92] [93] |
| Spectroscopic Method Validation | Multiple detection limits (LLD, ILD, CMDL, LOD, LOQ), matrix effects | Accuracy, recovery, calibration linearity, precision | Elemental analysis in alloys (e.g., Ag-Cu systems) [94] |
| Clinical Method Validation | Eight-step process comparing new vs. established methods | Accuracy, precision, reportable range, correlation | Diagnostic testing (e.g., hemoglobin A1C) [95] |
Experimental Protocols for Multi-Method Coordination Analysis
X-ray Photoelectron Diffraction (XPD) with DFT Calculations
The correlative XPD-DFT protocol for borophene structure determination exemplifies rigorous multi-method validation for two-dimensional materials [89]. The experimental workflow begins with the synthesis of the β12 borophene phase on Ag(111) substrates under ultra-high vacuum conditions. High-resolution core-level photoelectron spectroscopy measurements are then performed using synchrotron radiation sources, collecting B 1s spectra with high energy resolution. Subsequent X-ray photoelectron diffraction patterns are acquired by measuring the intensity variations of B 1s photoelectrons as a function of emission angle.
The parallel computational component employs density functional theory (DFT) calculations to model the electronic structure and atomic geometry of the borophene system. The critical validation step involves correlating the experimentally observed B 1s spectral features with the calculated electronic structures of non-equivalent boron atoms in the β12 unit cell. Specifically, the coordination number of each boron atom manifests in distinct binding energy signatures, allowing the experimental spectra to be deconvoluted into contributions from atoms with different coordination environments [89]. The XPD patterns provide direct structural validation through comparison with simulated diffraction patterns from the DFT-optimized structures, confirming the minimal corrugation of borophene on the Ag(111) substrate.
Gas-Phase Cluster Modeling with Mass Spectrometry
The gas-phase cluster approach enables atomic-level resolution of coordination effects by studying precisely-defined molecular analogs of catalytic sites [57]. The experimental apparatus centers on a modified linear ion trap mass spectrometer (LTQ-XL MS) equipped with an electrospray ionization (ESI) source. Coordination complexes are generated by electrospraying methanol solutions containing Cu(NO3)2 and selected nitrogen-donor ligands (pyridine, bipyridine, terpyridine, carbazole, acridine) or heteroatom-doped analogs.
The isolated Cu(I) complexes, representing well-defined coordination environments with specific N coordination numbers (1-4) and geometries (pyridinic, pyrrolic), are mass-selected and reacted with controlled introductions of CO gas. The reaction kinetics are monitored in real-time by tracking the formation of CO-adduct complexes, with pseudo-first-order rate constants (k) calculated from the temporal decay of precursor ions. High-resolution mass spectrometry (Q Exactive MS) confirms the exact elemental composition of all complexes.
Complementary density functional theory calculations provide the structural and electronic basis for interpreting the experimental kinetic data. Geometry optimization reveals bond lengths and coordination geometries, while population analysis examines charge transfer effects. Frontier orbital theory elucidates the electronic factors governing CO binding affinity across different coordination environments [57]. This combined experimental-theoretical approach establishes general rules for how coordination number and geometry regulate substrate binding in single-atom catalysts.
Figure 1: Workflow for Correlative Multi-Method Validation of Coordination Numbers
Quantitative Performance Comparison of Method Combinations
Detection Capabilities and Accuracy Metrics
Table 3: Analytical Performance of Validated Method Combinations
| Method Combination | Detection Limits | Precision (RSD) | Accuracy/Recovery | Coordination Number Range |
|---|---|---|---|---|
| HPLC-PDA (Metronidazole) | LOD: 0.02 µg/mL, LOQ: 0.05 µg/mL [93] | Injection repeatability: 0.5% (assay) [93] | Linear range: 75-225 µg/mL (r²=0.9999) [93] | Not applicable (molecular compound) |
| Spectroscopic (Ag-Cu Alloys) | LOD/LOQ matrix-dependent; CMDL at 95% confidence [94] | Demonstrated through repeated measurements | Recovery assessments confirm reliability [94] | Varies with composition and structure |
| XPD-DFT (Borophene) | Atomic coordination sensitivity demonstrated [89] | Spectral deconvolution precision established | Confirmed via diffraction pattern simulation [89] | Differentiated 3-7 coordination in β12 phase [89] |
| Gas-Phase Clusters (Cu-N-C) | Quantitative rate constants for CO binding [57] | Kinetic measurements across multiple replicates | Correlation with DFT calculations [57] | Systematically tested CN 0-4 [57] |
| MD Simulations (Bivalent Ions) | Coordination fluctuations < 0.1 across solvents [54] | Averaged over 16 independent trajectories | Agreement with experimental crystal data [54] | Mg²⁺: 6, Ca²⁺: 8, Be²⁺: 4 [54] |
Application-Specific Validation Criteria
Different scientific disciplines employ specialized validation criteria tailored to their specific accuracy requirements and operational constraints. In pharmaceutical analysis, method validation emphasizes strict regulatory compliance with demonstrated specificity, accuracy, precision, linearity, range, detection limits, quantification limits, robustness, and system suitability [92] [93]. For example, the validation of an HPLC method for furosemide quantification established linearity with R² > 0.995, accuracy recoveries of 98.2-101.0%, and precision with RSD ≤ 2% across intra-day and inter-day measurements [92].
In clinical diagnostics, the eight-step validation framework requires comparison against existing methods with defined performance thresholds [95]. For hemoglobin A1C analysis, validation demands demonstration of statistical identity between methods (slope = 1.00 within 95% confidence, intercept = 0.00 within 95% confidence), with 100% of results falling within the total allowable error budget [95].
For coordination number determination in materials science, validation typically involves cross-method verification where multiple techniques converge on consistent coordination assignments. The topological coordination number (tCN) approach provides a sophisticated validation framework through its incorporation of coordination reciprocity requirements and ranking of different sub-coordination scenarios by relative weights [15]. This enables more nuanced characterization of complex structures where multiple coordination environments may coexist with similar probabilities.
Research Reagent Solutions for Coordination Studies
Table 4: Essential Materials and Reagents for Coordination Analysis Experiments
| Reagent/Instrument Category | Specific Examples | Function in Coordination Analysis | Application Context |
|---|---|---|---|
| Chromatography Columns | Water Symmetry C18 (100 mm × 4.6 mm, 3.5 μm) [93] | Separation of analytes and degradation products | Pharmaceutical compound analysis [93] |
| HPLC Solvents & Mobile Phases | Isopropyl alcohol/water (20:80 v/v) [93]; 0.1% acetic acid in water/acetonitrile [92] | Mobile phase for compound separation | Greener analytical methods for pharmaceuticals [93] |
| Reference Materials | Ag-Cu alloys (x = 0.05, 0.1, 0.3, 0.75, 0.9) [94] | Calibration standards for spectroscopic analysis | Matrix effect studies in elemental analysis [94] |
| Coordination Complex Ligands | Pyridine, bipyridine, terpyridine, carbazole, acridine [57] | Modeling specific coordination environments | Gas-phase cluster studies of single-atom catalysts [57] |
| Computational Chemistry Software | GROMACS [54], DFT packages | Molecular dynamics and electronic structure calculation | Theoretical validation of coordination numbers [54] |
| Crystalline Substrates | Ag(111) single crystal substrates [89] | Epitaxial growth template for 2D materials | Borophene structure determination [89] |
Figure 2: Coordination Environments and Analysis Approaches
The systematic comparison of validation approaches demonstrates that correlative multi-method strategies significantly enhance the reliability and accuracy of coordination number determination across diverse scientific domains. By integrating complementary techniques that probe coordination environments through different physical principles—such as combining X-ray photoelectron diffraction with DFT calculations [89], or gas-phase cluster models with kinetic measurements and theoretical analysis [57]—researchers can overcome the limitations inherent in any single methodological approach.
The most effective validation frameworks incorporate both quantitative statistical measures and qualitative assessment criteria [90] [91], establishing congruence, convergence, and credibility through triangulation of evidence from multiple sources. As coordination number analysis increasingly informs critical applications in catalyst design, battery development, and pharmaceutical sciences, the adoption of robust multi-method validation protocols will be essential for generating reliable structure-property relationships and advancing materials innovation.
Future methodological developments will likely focus on standardizing validation criteria across disciplines, improving computational-experimental integration, and developing automated validation workflows that can efficiently handle the complex coordination scenarios encountered in advanced functional materials. Through continued refinement of these correlative multi-method approaches, the scientific community can address existing challenges in coordination number determination and unlock new opportunities for tailored material design across chemical, energy, and biomedical applications.
In heterogeneous catalysis, the coordination number (CN), defined as the number of nearest neighbor atoms to a specific metal atom, serves as a fundamental geometric descriptor that directly influences catalytic activity and selectivity. The ability to precisely control and measure the coordination environment of active sites, from single atoms and sub-nanometer clusters to nanoparticles, has opened new avenues for establishing atomic-level structure–performance relationships. This guide synthesizes current research to objectively compare how coordination number manipulation impacts catalytic function across various metals, support materials, and reaction systems, providing researchers with validated experimental protocols and data for designing next-generation catalysts.
Theoretical Foundation: Coordination Number as a Activity Descriptor
The coordination number of a catalytic active site profoundly affects its electronic structure and, consequently, its binding strength with reaction intermediates. Generalized Coordination Numbers (GCN) extend this concept by accounting for the coordination environment of neighboring atoms, providing a more sophisticated descriptor that correlates well with adsorption energies on both extended surfaces and nanoparticles [50].
The underlying principle is that as the coordination number of a metal atom decreases, the atom becomes increasingly under-coordinated and chemically unsaturated. This electron deficiency enhances its ability to form strong bonds with adsorbates. For face-centered cubic (fcc) metals, coordination numbers theoretically range from 12 (fully coordinated bulk atoms) to values as low as 6 or even lower for corner atoms on nanoparticles [96] [50]. This relationship creates opportunities to tune catalytic properties by strategically designing sites with specific coordination environments.
Table 1: Common Coordination Environments in Face-Centered Cubic (fcc) Nanostructures
| Site Location | Traditional CN | Generalized CN (ȲCN) | Typical Properties |
|---|---|---|---|
| Bulk Terrace | 9-12 | 9-12 | Saturated, weak adsorbate binding |
| Flat Terrace (111) | 9 | ~7.0-7.3 | Moderate activity |
| Step Edge | 7 | ~6.3-6.5 | High activity |
| Corner | 6-7 | <6.0 | Very high activity, often optimal |
Quantitative Impact on Catalytic Activity
Gold Nanoparticles for Hydrogen Isotope Exchange
A definitive study on gold nanoparticles supported on γ-Al₂O₃ demonstrated dramatic activity differences between low-coordination atoms. Researchers synthesized nanoparticles ranging from 0.7 to 40 nm and characterized them using HRTEM, SEM, XRD, DLS, and UV-Vis spectroscopy [96]. The specific activity for the H₂ + D₂ 2HD isotope exchange reaction at 77 K increased sharply when particle sizes decreased below 3 nm, where the population of low-coordination atoms becomes significant [96].
Critical findings from this research revealed:
- Corner atoms (CN = 6) exhibited a turnover frequency (TOF) of 0.258 ± 0.020 molecules site⁻¹ s⁻¹
- Edge atoms (CN = 7) showed a TOF of only 0.006 ± 0.001 molecules site⁻¹ s⁻¹
- This represents a more than 40-fold activity enhancement for corner versus edge atoms [96]
The experimental data clearly demonstrates that catalytic activity increases significantly when particle size decreases below 3 nm, with the most dramatic enhancement occurring below 4 nm where the proportion of very low-coordination sites increases exponentially [96].
Table 2: Catalytic Activity of Gold Nanoparticles of Different Sizes for Hydrogen Isotope Exchange
| Particle Size (nm) | logKₛₚ (Molecules s⁻¹ cm⁻²) | Activation Energy HT (kJ mol⁻¹) | Activation Energy LT (kJ mol⁻¹) |
|---|---|---|---|
| 0.7 ± 0.2 | 14.37 | 9.6 ± 2.1 | 0.16 ± 0.39 |
| 4.6 ± 0.8 | 13.1 | 8.5 ± 1.9 | -0.69 ± 0.92 |
| 14.4 ± 2.2 | 12.19 | 23.4 ± 8.8 | 1.7 ± 0.6 |
| 28.3 ± 3.1 | 11.93 | 42.1 ± 9.3 | 1.5 ± 0.4 |
| 40.1 ± 5.4 | 11.48 | 36 ± 13.3 | 2.0 ± 0.7 |
Platinum-Based Systems for Dehydrogenation and Hydrogenation
n-Butane Dehydrogenation (DDH)
A systematic study constructed a relationship between the average Pt-Pt coordination number and n-butane DDH performance using Pt species supported on defective nanodiamond@graphene (ND@G) [97]. Researchers fabricated fully exposed Pt₃ clusters stabilized by atomically dispersed Sn promoters, achieving precise control over Pt coordination environments.
Performance comparison revealed:
- Fully exposed Pt₃ clusters demonstrated superior performance with 35.4% conversion and 99.0% alkene selectivity at 450°C
- This performance exceeded both Pt nanoparticles and Pt single atoms
- Density functional theory (DFT) calculations attributed the high activity to favorable dehydrogenation activation barriers and reasonable desorption barriers of butene [97]
The study established a clear structure-performance relationship, highlighting that moderate coordination numbers in fully exposed clusters provide the optimal balance between reactant activation and product desorption.
Hydrogenation Reactions
Research on Pt single-atom catalysts (SACs) supported on Fe₂O₃ demonstrated that coordination number tuning directly impacts hydrogenation activity [98]. Using an ethanediamine-assisted rapid thermal treatment (en-M-RTT) method, scientists prepared a series of Pt₁/Fe₂O₃-T catalysts with varying Pt-O coordination numbers by adjusting the RTT temperature from 500 to 600°C [98].
Key observations included:
- As the Pt-O coordination number decreased, the oxidation state of Pt decreased correspondingly
- This reduction in coordination number and oxidation state led to a systematic increase in hydrogenation activity
- The tunability demonstrated that SACs can bridge the gap between heterogeneous and homogeneous catalysis, where ligand effects similarly modulate activity [98]
Impact on Catalytic Selectivity
The coordination environment of active sites not only affects activity but also plays a determining role in reaction selectivity. Lower coordination sites often favor deeper activation of molecules, which can influence reaction pathways and product distributions.
Methane Oxidation on Platinum
A structure-descriptor-based microkinetic model for complete methane oxidation on Pt nanoparticles revealed a volcano-like relationship between coordination number and reaction rate [99]. Contrary to conventional expectations that smaller particles always show higher activity, the model predicted an optimal coordination number for maximum activity [99].
Interestingly, the study found that:
- Very small particles exhibited low reactivity due to carbon poisoning
- The variation in experimental literature data could be largely reconciled by considering the structure sensitivity of the reaction and different coordination environments present in various catalyst preparations [99]
This demonstrates that selectivity between complete combustion and partial oxidation, or competing degradation pathways, can be controlled by designing catalysts with specific coordination environments.
Experimental Protocols for Coordination Number Determination and Catalyst Evaluation
Objective: Prepare gold nanoparticles with controlled sizes (0.7-40 nm) on γ-Al₂O₃ support to study size-dependent coordination effects.
Materials:
- Gold precursor (typically HAuCl₄·3H₂O)
- γ-Al₂O₃ support
- Appropriate reducing/stabilizing agents (e.g., sodium citrate, NaBH₄)
- Deionized water and solvents
Procedure:
- Synthesize colloidal gold nanoparticles of specific sizes by varying reduction conditions
- Deposit nanoparticles onto γ-Al₂O₃ support via impregnation or deposition-precipitation
- Dry catalyst at moderate temperature (e.g., 100°C)
- Calcine below 573 K to prevent nanoparticle coalescence [96]
Characterization:
- HRTEM: Determine particle size distribution and morphology
- XRD: Confirm crystalline structure and estimate size via Scherrer equation
- DLS: Measure hydrodynamic size in solution
- UV-Vis Spectroscopy: Characterize surface plasmon resonance changes
Objective: Evaluate catalytic activity of different coordination sites for H₂ + D₂ 2HD exchange.
Experimental Conditions:
- Temperature: 77 K (to suppress other thermally activated reactions)
- Reactant mixture: H₂ + D₂ in specific ratio
- Catalyst mass: Adjusted to maintain differential conversion conditions
Activity Measurement:
- Place catalyst in reaction chamber and cool to 77 K
- Introduce H₂/D₂ mixture
- Monitor HD formation over time using mass spectrometry
- Calculate specific activity (Ksp) using: Ksp = K₀ × NT / SH Where K₀ = first-order rate constant, NT = number of hydrogen atoms, SH = active surface area
- Determine TOF for specific sites based on coordination number assignment
Objective: Prepare oxide-supported single-atom catalysts with tunable coordination numbers.
Materials:
- Metal precursor (e.g., Pt(NH₃)₄(NO₃)₂)
- Ethanediamine (en) ligand
- Support material (e.g., Fe₂O₃, Al₂O₃)
- Inert gas (He or N₂) for thermal treatment
Procedure:
- Chelate Pt cations with ethanediamine to form [Pt(en)₂]²⁺ complex
- Impregnate complex onto support material
- Dry at moderate temperature
- Apply Rapid Thermal Treatment (RTT) in He atmosphere:
- Temperature range: 500-600°C
- Duration: 1 minute (critical to prevent aggregation)
- Rapid heating and cooling rates [98]
Characterization:
- AC-HAADF-STEM: Confirm single-atom dispersion
- EXAFS: Determine Pt-O coordination number and absence of Pt-Pt bonds
- XPS: Monitor oxidation state changes
Data Visualization: Relationships and Workflows
Coordination Number Impact Pathway
SAC Synthesis with Tunable CN
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Essential Research Reagents and Materials for Coordination Number Studies
| Reagent/Material | Function/Purpose | Example Application |
|---|---|---|
| Metal Precursors (e.g., HAuCl₄, Pt(NH₃)₄(NO₃)₂) | Source of catalytic metal species | Synthesis of Au nanoparticles [96], Pt single-atoms [98] |
| Ethanediamine (en) Ligand | Chelating agent for metal cations | Preventing aggregation in SAC synthesis [98] |
| High-Surface-Area Supports (γ-Al₂O₃, Fe₂O₃, ND@G) | Stabilize metal species, provide anchoring sites | Supporting Au nanoparticles [96], Pt single-atoms [98] |
| Sn Promoter | Geometric partitioning of Pt atoms | Fabrication of fully exposed Pt₃ clusters [97] |
| Inert Gas Supply (He, N₂) | Atmosphere for thermal treatments | RTT process for SAC synthesis [98] |
The coordination number serves as a powerful descriptor and design parameter for tailoring catalytic performance. Experimental evidence across multiple systems consistently demonstrates that low-coordination sites (CN = 6-7) can exhibit order-of-magnitude higher activity than their higher-coordination counterparts, while intermediate coordination environments often optimize selectivity by balancing activation and desorption processes. The development of sophisticated synthesis methods, particularly the en-M-RTT approach for single-atom catalysts and promoter-stabilized clusters, now enables precise control over coordination environments. For researchers pursuing catalyst development, focusing on coordination number as a fundamental design parameter provides a rational pathway to enhanced catalytic performance across diverse applications including hydrogenation, dehydrogenation, and oxidation catalysis.
Emerging Standards and Best Practices for Method Validation
The field of analytical method validation is undergoing a significant transformation, shifting from a prescriptive, "check-the-box" approach to a more scientific, risk-based, and lifecycle-oriented model. This evolution is largely driven by updated international guidelines and the increasing complexity of analytical techniques, particularly for sophisticated applications like coordination number determination in pharmaceutical research. For researchers, scientists, and drug development professionals, understanding these changes is critical for ensuring regulatory compliance, data integrity, and the reliability of research outcomes.
The simultaneous release of the revised ICH Q2(R2) guideline on the validation of analytical procedures and the new ICH Q14 guideline on analytical procedure development represents a fundamental modernization of the framework for analytical methods [100]. This update expands the scope to include modern technologies and explicitly encourages a more systematic, knowledge-rich approach to development and validation. Furthermore, the U.S. Food and Drug Administration (FDA), as a key member of the International Council for Harmonisation (ICH), adopts and implements these harmonized guidelines, making compliance with ICH standards a direct path to meeting FDA requirements for submissions such as New Drug Applications (NDAs) and Abbreviated New Drug Applications (ANDAs) [100]. This article will compare traditional and emerging methodologies, provide supporting experimental data, and detail best practices for validating methods within the context of cutting-edge research, such as evaluating the accuracy of coordination number determination techniques.
Core Principles: Method Validation Parameters and Lifecycle Management
Fundamental Validation Parameters
At its core, method validation demonstrates that a procedure is fit for its intended purpose. ICH Q2(R2) outlines a set of fundamental performance characteristics that must be evaluated, the rigor of which depends on the type of method (e.g., quantitative, qualitative) [100].
The table below summarizes the key validation parameters and their definitions:
Table 1: Core Analytical Method Validation Parameters as per ICH Q2(R2)
| Parameter | Definition |
|---|---|
| Accuracy | The closeness of agreement between the test result and the true value [100]. |
| Precision | The degree of agreement among individual test results when the procedure is applied repeatedly to multiple samplings of a homogeneous sample. This includes repeatability, intermediate precision, and reproducibility [100]. |
| Specificity | The ability to assess the analyte unequivocally in the presence of components that may be expected to be present, such as impurities, degradation products, or matrix components [100]. |
| Linearity | The ability of the method to elicit test results that are directly proportional to the concentration of the analyte within a given range [100]. |
| Range | The interval between the upper and lower concentrations of the analyte for which the method has demonstrated suitable linearity, accuracy, and precision [100]. |
| Limit of Detection (LOD) | The lowest amount of analyte in a sample that can be detected, but not necessarily quantitated [100]. |
| Limit of Quantitation (LOQ) | The lowest amount of analyte in a sample that can be determined with acceptable accuracy and precision [100]. |
| Robustness | A measure of the method's capacity to remain unaffected by small, deliberate variations in method parameters, indicating its reliability during normal usage [100]. |
The Lifecycle Approach: ICH Q2(R2) and Q14
The most significant modernization introduced by the latest ICH guidelines is the concept of an Analytical Procedure Lifecycle. This moves validation from a one-time event to a continuous process that begins with method development and continues through routine use and post-approval change management [100].
Central to this enhanced approach is the Analytical Target Profile (ATP). Introduced in ICH Q14, the ATP is a prospective summary of the intended purpose of the analytical procedure and its required performance criteria [100]. By defining the ATP at the outset—for instance, specifying the required precision and accuracy for a coordination number measurement—a laboratory can design a fit-for-purpose method and a validation plan that directly addresses its specific needs. This science- and risk-based foundation allows for more flexible and efficient management of changes throughout the method's lifecycle.
Experimental Protocols for Method Validation
A critical experiment in method validation, particularly for assessing the systematic error or inaccuracy of a new method (the "test method"), is the Comparison of Methods experiment [101].
Comparison of Methods Experiment
Purpose: To estimate the inaccuracy or systematic error of a test method by comparing its results against those from a validated comparative method using real patient specimens [101].
Experimental Design and Protocol:
- Selection of Comparative Method: The ideal comparative method is a "reference method" whose correctness is well-documented. When using a routine method for comparison, differences must be interpreted carefully, as it may be unclear which method is responsible for any observed discrepancy [101].
- Specimen Selection and Number: A minimum of 40 different patient specimens is recommended. These specimens should be carefully selected to cover the entire working range of the method and should represent the spectrum of diseases and matrices expected in routine application. The quality and range of specimens are more critical than a large number of random specimens [101].
- Replication and Timeframe: Analyses should be performed over a minimum of 5 days, and ideally 20 days, to account for run-to-run variability. While single measurements are common, performing duplicate measurements by the test and comparative methods helps identify sample mix-ups or transposition errors [101].
- Specimen Handling: Specimens should be analyzed within two hours of each other by both methods to prevent changes in the specimen from being misinterpreted as analytical error. Stability requirements should be defined and systematized prior to the study [101].
Data Analysis and Statistical Evaluation
The analysis of comparison data involves both graphical and statistical techniques [101].
- Graphical Analysis: The data should be plotted to visualize the relationship and identify discrepancies.
- A Difference Plot (test result minus comparative result vs. comparative result) is used when methods are expected to agree one-to-one. The differences should scatter randomly around zero.
- A Comparison Plot (test result vs. comparative result) is used when methods are not expected to show one-to-one agreement. A visual line of best fit is drawn to show the general relationship.
- Statistical Calculations: For data covering a wide analytical range, linear regression analysis is preferred. It provides the slope and y-intercept of the line of best fit, which can be used to estimate the systematic error at any critical medical decision concentration (Xc). The systematic error (SE) is calculated as SE = Yc - Xc, where Yc is the value predicted by the regression line (Yc = a + bXc) [101]. The correlation coefficient (r) is also calculated, but it is more useful for verifying a sufficient data range (r ≥ 0.99) than for judging method acceptability [101].
Table 2: Key Phases in the Analytical Method Lifecycle
| Lifecycle Phase | Key Activities | Guideline Reference |
|---|---|---|
| Procedure Design & Development | Define the Analytical Target Profile (ATP); apply Quality by Design (QbD) principles; select method type; conduct risk assessments. | ICH Q14 [100] |
| Procedure Performance Qualification (Validation) | Conduct formal validation studies to demonstrate the method meets the ATP criteria for parameters like accuracy, precision, and specificity. | ICH Q2(R2) [100] |
| Continued Procedure Performance Verification | Ongoing monitoring of method performance during routine use; manage post-approval changes through a structured, science-based change management process. | ICH Q12 [100] |
The Scientist's Toolkit: Essential Research Reagent Solutions
The reliability of any validated method hinges on the quality of the materials used. The following table details essential reagents and their functions in ensuring accurate and reproducible results, particularly in complex analyses.
Table 3: Essential Research Reagents and Materials for Method Validation
| Reagent/Material | Function and Importance in Validation |
|---|---|
| Certified Reference Materials (CRMs) | A CRM is a reference material characterized by a metrologically valid procedure, accompanied by a certificate that provides the value of the specified property, its associated uncertainty, and a statement of metrological traceability. They are vital for assessing the accuracy, precision, and sensitivity of analytical measurements and for method validation [102]. |
| Matrix-Based Reference Materials | These are RMs that mimic the sample matrix (e.g., plant material, liquid extract). They are essential for addressing analytical challenges like accounting for extraction efficiency and the effect of interfering compounds, thereby demonstrating that a method is "fit-for-purpose" for a specific matrix [102]. |
| Color Reference Solutions | Ready-to-use solutions, such as those defined in the European Pharmacopoeia, are used as calibration and quality control standards to measure the degree of coloration of liquids, which can be a critical quality attribute [103]. |
| Stable Isotope-Labeled Internal Standards | Used particularly in mass spectrometry, these are added to samples to correct for variability in sample preparation, injection, and ionization efficiency, thereby significantly improving the precision and accuracy of quantitative results. |
| System Suitability Test Solutions | Mixtures of specific analytes used to verify that the entire analytical system (from instrument to reagents to columns) is performing adequately at the start of, and during, an analytical sequence. |
Method Comparison Workflow and Coordination Number Analysis
The following diagram illustrates the logical workflow for a method comparison experiment, from planning to final interpretation, which can be adapted for various validation studies.
Method Comparison Experiment Workflow
Application to Coordination Number Determination Research
In research focused on evaluating the accuracy of coordination number determination methods, the principles of method validation are paramount. For instance, in a coupled DEM-DFN (Discrete Element Model-Discrete Fracture Network) model used to simulate fractured rocks, the coordination number is a crucial microstructural index that characterizes the connection of microstructural elements and helps identify crack patterns in spatial distribution [104].
Validating a new computational or analytical method for determining coordination numbers against an established method (e.g., high-resolution digital imaging) would directly employ the comparison of methods experiment. The "test method" would be the new computational technique, while the "comparative method" would be the imaging analysis. The "specimens" would be different rock models or samples with a known or variably fractured structure. The systematic error estimated from the regression analysis would provide a quantitative measure of the new method's accuracy (bias) in determining coordination numbers across a range of fracture densities.
The emerging standards for method validation, championed by ICH Q2(R2) and Q14, represent a paradigm shift toward a more scientific, proactive, and lifecycle-based approach. The adoption of concepts like the Analytical Target Profile, enhanced development practices, and a risk-based control strategy empowers researchers to build quality into their methods from the very beginning. For scientists engaged in advanced research, such as developing and validating novel methods for coordination number determination, adhering to these best practices—rigorous experimental design, statistical evaluation of comparative data, and the use of high-quality reference materials—is non-negotiable. It ensures the generation of reliable, reproducible, and defensible data that can accelerate drug development and meet the stringent demands of global regulatory bodies.
Conclusion
The accurate determination of coordination numbers requires careful method selection tailored to specific chemical environments and research objectives. While traditional geometric approaches provide valuable initial insights, advanced techniques based on electron density topology and spectroscopy offer superior accuracy, especially for complex systems with dynamic coordination environments or significant atomic size disparities. The integration of multiple complementary methods—combining experimental spectroscopy with computational modeling and machine learning analysis—represents the most robust approach for validation. Future directions will likely focus on real-time coordination monitoring, automated symmetry analysis, and AI-driven prediction of coordination-dependent properties, with significant implications for rational drug design, metalloprotein engineering, and the development of advanced catalytic systems for pharmaceutical synthesis. The ongoing refinement of coordination number determination methods will continue to provide fundamental insights driving innovation across biomedical and clinical research.
References
- 1. (PDF) Coordination in liquid argon number [https://www.academia.edu/12182482/Coordination_number_in_liquid_argon]
- 2. A validated experimental NMR parameter dataset of organic... [https://pubs.rsc.org/en/content/articlelanding/2025/an/d5an00240k]
- 3. Visualization of very large high-dimensional data sets as ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-020-0416-x]
- 4. Topological coordination numbers and ... - IUCr Journals [https://journals.iucr.org/paper?pen5011]
- 5. Article Uncovering electronic and geometric descriptors of ... [https://www.sciencedirect.com/science/article/pii/S2667109321001457]
- 6. A Survey of Quantitative Descriptions of Molecular Structure [https://pmc.ncbi.nlm.nih.gov/articles/PMC3809149/]
- 7. An improved dataset of force fields, electronic and ... [https://www.nature.com/articles/s41597-024-03707-0]
- 8. Correlative analysis of Ni(II) coordination states in molten salts ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc01059d]
- 9. Comparison of Descriptor- and Fingerprint Sets in Machine ... [https://www.frontiersin.org/journals/chemistry/articles/10.3389/fchem.2022.852893/full]
- 10. Graph neural networks for predicting metal–ligand ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12541316/]
- 11. Oxidation states in solids from data-driven paradigms [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc05694b]
- 12. On the Relationship and Distinction Between Atomic ... [https://arxiv.org/html/2508.16808v1]
- 13. A Comparison of Experimental and Ab Initio Structural Data ... [https://www.mdpi.com/2075-4701/13/6/1096]
- 14. Correlative analysis of Ni(ii) coordination states in molten ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12063551/]
- 15. Topological coordination numbers and ... - IUCr Journals [https://journals.iucr.org/a/issues/2025/03/00/pen5011/]
- 16. Coordination number [https://en.wikipedia.org/wiki/Coordination_number]
- 17. Intermetallics 2025 [https://intermetallics-conference.de/]
- 18. Sub-angstrom strain in high-entropy intermetallic boosts ... [https://www.nature.com/articles/s41467-025-62725-7]
- 19. Coordination-in-pipe engineering of Pt-based intermetallic ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d4sc07905a]
- 20. Coordination Chemistry Reviews | Vol 522, 1 January 2025 [https://www.sciencedirect.com/journal/coordination-chemistry-reviews/vol/522/suppl/C]
- 21. Cu/Zn/histidine supramolecular assemblies with optimized ... [https://www.nature.com/articles/s41467-025-65074-7]
- 22. Field-dependent relaxation profiles of biomolecular systems [https://pubs.rsc.org/en/content/articlehtml/2025/cp/d4cp04306e]
- 23. CAVD, towards better characterization of void space for ... [https://www.nature.com/articles/s41597-020-0491-x]
- 24. Focus on Quantum Crystallography - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC12573928/]
- 25. Boosting heterogeneous catalyst discovery by structurally ... [https://www.sciencedirect.com/science/article/pii/S2468519423001684]
- 26. Residual point generation methods using Voronoi ... [https://www.sciencedirect.com/science/article/pii/S002199912500405X]
- 27. Topological coordination numbers and ... [https://ui.adsabs.harvard.edu/abs/2025AcCrA..81..221W/abstract]
- 28. electron-deficient multicenter bonds in electron-rich elements [https://pubs.rsc.org/en/content/articlehtml/2025/tc/d5tc02328a]
- 29. A Topological Analysis of Electron Density and Band ... [https://www.mdpi.com/1422-0067/3/1/38]
- 30. Accelerating discovery of glass materials in electronic devices ... [https://pubs.rsc.org/en/content/articlehtml/2025/ta/d4ta08686d]
- 31. Accelerated Discovery of Topological Conductors for ... [https://arxiv.org/html/2509.15135v1]
- 32. The concept of 'color' topological type: classification and ... [https://pubs.rsc.org/en/content/articlehtml/2025/ce/d5ce00590f]
- 33. Fundamentals of XAS Data Analysis: A Hands-on Tutorial ... [https://www.bnl.gov/xascourse/]
- 34. X-Ray Absorption and Emission Spectroscopy in ... [https://www.mdpi.com/2076-3417/15/19/10784]
- 35. In situ X-ray spectroscopies beyond conventional X ... [https://www.nature.com/articles/s41467-023-42370-8]
- 36. X-ray Absorption Spectroscopy: Introduction to Experimental ... [https://www-ssrl.slac.stanford.edu/mes/xafs/xas_intro.html]
- 37. Review Progress in the theory and interpretation of XANES [https://www.sciencedirect.com/science/article/abs/pii/S0010854504000414]
- 38. Spectra-to-Structure and Structure-to-Spectra Inference ... [https://arxiv.org/html/2506.11908v1]
- 39. X-ray Absorption Spectroscopy (XAS) Data Analysis ... [https://www.diamond.ac.uk/Home/Events/2025/XAS-Workshop-2025.html]
- 40. Article Random Forest Models for Accurate Identification of ... [https://www.sciencedirect.com/science/article/pii/S2666389920300131]
- 41. (IUCr) A new framework for X-ray absorption spectroscopy ... [https://journals.iucr.org/s/issues/2025/05/00/rv5192/]
- 42. XAS applied to pharmaceuticals: drug administration and ... [https://pubmed.ncbi.nlm.nih.gov/11513002/]
- 43. CSM Software: Continuous Symmetry and Chirality ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11267602/]
- 44. Determining molecular structure, coordination geometry ... [https://www.nature.com/articles/s41467-025-66054-7]
- 45. Continuous Symmetry Discovery and Enforcement Using ... [https://arxiv.org/html/2505.08219v2]
- 46. Investigating the effect of active site's coordination number ... [https://www.sciencedirect.com/science/article/abs/pii/S0360319925006895]
- 47. Unraveling the local coordination effect of Cu–N–C single ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12123374/]
- 48. Comparisons of particle clustering phenomenon between ... [https://www.sciencedirect.com/science/article/abs/pii/S003259102101007X]
- 49. Structures and Reactivity of Isolated Metal Clusters [https://www.int.kit.edu/126.php]
- 50. The ABC of Generalized Coordination Numbers and Their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10369287/]
- 51. Magnetic and structural properties of isolated and ... [https://www.sciencedirect.com/science/article/abs/pii/S0167572904001001]
- 52. DFT Calculation of Physical Properties for Performance ... [https://ui.adsabs.harvard.edu/abs/2025PhSS...67..821B/abstract]
- 53. Ab initio characterization of protein molecular dynamics ... [https://www.nature.com/articles/s41586-024-08127-z]
- 54. Coordination Numbers of Bivalent Ions in Organic Solvents [https://link.springer.com/article/10.1134/S0036024421100204]
- 55. Coordination Number - an overview [https://www.sciencedirect.com/topics/chemistry/coordination-number]
- 56. CryoSIM: super-resolution 3D structured illumination ... [https://opg.optica.org/optica/abstract.cfm?uri=optica-7-7-802]
- 57. Unraveling the local coordination effect of Cu–N–C single ... [https://pubs.rsc.org/en/content/articlehtml/2025/sc/d5sc01982f]
- 58. A templating approach with phase change to tailored ... [https://www.nature.com/articles/s41467-025-63117-7]
- 59. Ligand-performance relationships of surface-modified CeO ... [https://www.sciencedirect.com/science/article/abs/pii/S2468023025019868]
- 60. An investigation of the coordination chemistry ... [https://www.sciencedirect.com/science/article/abs/pii/S0020169322003668]
- 61. Comprehensive coordination chemistry of ... [https://pubs.rsc.org/en/content/articlehtml/2025/qi/d5qi00275c]
- 62. Successive modification of polydentate complexes gives ... [https://www.nature.com/articles/s41467-019-09367-8]
- 63. a comparative study of the copper(II) and zinc(II) coordination ... [https://pubs.rsc.org/en/content/articlehtml/2025/nr/d5nr01087j]
- 64. Graph neural networks for predicting metal-ligand ... [https://pubmed.ncbi.nlm.nih.gov/41052327/]
- 65. Spectroscopic Analysis | Research Starters [https://www.ebsco.com/research-starters/chemistry/spectroscopic-analysis]
- 66. Staying Updated with Spectroscopic Techniques [https://www.spectroscopyonline.com/view/staying-updated-with-spectroscopic-techniques-how-lead-investigators-adapt-to-a-changing-industry]
- 67. Spectroscopy Technique - an overview [https://www.sciencedirect.com/topics/materials-science/spectroscopy-technique]
- 68. Comparison of spectroscopic techniques for determination ... [https://www.sciencedirect.com/science/article/pii/S1386142525006511]
- 69. Comparing Spectroscopic Techniques for Nanoscale ... [https://www.azonano.com/article.aspx?ArticleID=6934]
- 70. Comparison of Spectroscopic Techniques Using the ... [https://link.springer.com/article/10.1007/s12161-023-02568-4]
- 71. Quantitative analysis of EXAFS data sets using deep ... [https://www.nature.com/articles/s41598-025-94376-5]
- 72. Machine learning for analysis of experimental scattering ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10718081/]
- 73. A new framework for X-ray absorption spectroscopy data ... [https://journals.iucr.org/paper?rv5192]
- 74. Multivariate curve resolution: A review of advanced and ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267012018351]
- 75. Multivariate curve resolution: a powerful tool for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC137437/]
- 76. Simultaneous Determination of Drugs Affecting Central Nervous... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7036114/]
- 77. Multivariate curve resolution for kinetic modeling and scale ... [https://link.springer.com/article/10.1007/s41981-022-00252-y]
- 78. Identification of Reliable Components in Multivariate Curve ... [https://www.nature.com/articles/srep15710]
- 79. Multivariate curve resolution-soft independent modelling of ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267024000060]
- 80. Strategies for robust, accurate, and generalizable ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12607264/]
- 81. Manipulating d-orbital of Cu single atom site by ... [https://www.nature.com/articles/s41467-025-61198-y]
- 82. Self-healing Cu single-atom catalyst for high-performance ... [https://www.nature.com/articles/s41467-025-63274-9]
- 83. What Is the Difference Between FCC and BCC? (Crystal Structure, Properties, Interstitial Sites, and Examples) | Materials Science & Engineering Student [https://msestudent.com/what-is-the-difference-between-fcc-and-bcc-crystal-structure-properties-interstitial-sites-and-examples/]
- 84. Cubic crystal system - Wikipedia [https://en.wikipedia.org/wiki/Cubic_crystal_system]
- 85. Comparison of SC, BCC, FCC, and HCP Crystal Structures | Materials Science & Engineering Student [https://msestudent.com/comparison-of-sc-bcc-fcc-and-hcp/]
- 86. crystals - Can I distinguish hexagonal close packing (HCP) from face centred cubic (FCC) arrangement based on Fourier transform - Physics Stack Exchange [https://physics.stackexchange.com/questions/579259/can-i-distinguish-hexagonal-close-packing-hcp-from-face-centred-cubic-fcc-ar]
- 87. Accurate simulation of surfaces and interfaces of ten FCC ... [https://www.nature.com/articles/s41524-020-00478-1]
- 88. Lattice distortions and non-sluggish diffusion in BCC ... [https://arxiv.org/html/2508.15558v1]
- 89. Determining the atomic coordination number in ... [https://www.sciencedirect.com/science/article/pii/S2468023024009489]
- 90. Mixed methods instrument validation: Evaluation ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9875772/]
- 91. A multi-dimensional approach to improve validation ... [https://www.sciencedirect.com/science/article/pii/S2666049024000331]
- 92. Simultaneous Development and Validation of an HPLC ... [https://www.mdpi.com/1420-3049/30/19/4031]
- 93. Development and validation of a HPLC–PDA method for ... [https://link.springer.com/article/10.1007/s44371-025-00109-y]
- 94. Validation of analytical methods and detection limits in ... [https://www.sciencedirect.com/science/article/abs/pii/S0969806X24009745]
- 95. Eight Steps to Method Validation in a Clinical Diagnostic ... [https://clsjournal.ascls.org/content/31/1/43]
- 96. Difference in the Catalytic Activity of Atoms in the Corners and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11594137/]
- 97. Regulating coordination number in atomically dispersed Pt ... [https://www.nature.com/articles/s41467-021-22948-w]
- 98. Unraveling the coordination structure-performance ... [https://www.nature.com/articles/s41467-019-12459-0]
- 99. Reconciling experimental catalytic data stemming from ... [https://pubs.rsc.org/en/content/articlehtml/2023/sc/d2sc06819b]
- 100. ICH and FDA Guidelines for Analytical Method Validation [https://www.labmanager.com/ich-and-fda-guidelines-for-analytical-method-validation-34252]
- 101. The Comparison of Methods Experiment [https://www.westgard.com/lessons/basic-method-validation/lesson23.html]
- 102. The Importance of Reference Materials and Method ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8713974/]
- 103. Color Reference Solutions analytical standard ... [https://www.sigmaaldrich.com/US/en/product/sial/87448?srsltid=AfmBOoqy8SWD8ky25ofJAHjMzQAqvL8__v4x8rXG6LtHA0xnbALFvKf4]
- 104. On the Determination of Coordination Numbers of Coupled ... [https://www.frontiersin.org/journals/earth-science/articles/10.3389/feart.2021.665275/full]