Achieving SCF Convergence in Computational Drug Discovery: A Comprehensive Guide to Parameter Setting and Optimization
This article provides a comprehensive guide for researchers and drug development professionals on setting and optimizing parameters to achieve Self-Consistent Field (SCF) convergence in computational chemistry calculations.
Achieving SCF Convergence in Computational Drug Discovery: A Comprehensive Guide to Parameter Setting and Optimization
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on setting and optimizing parameters to achieve Self-Consistent Field (SCF) convergence in computational chemistry calculations. Covering foundational principles, advanced methodological applications, systematic troubleshooting, and rigorous validation techniques, it addresses critical challenges in electronic structure calculations for biomolecular systems. By synthesizing current methodologies and optimization strategies, this guide aims to enhance the reliability and efficiency of quantum chemical computations in pharmaceutical research, ultimately accelerating the drug discovery pipeline.
Understanding SCF Convergence: Fundamental Principles and Challenges in Quantum Chemistry for Drug Discovery
The Critical Role of SCF Convergence in Accurate Molecular Property Prediction
The Self-Consistent Field (SCF) method is an iterative computational procedure central to quantum chemical calculations based on Density Functional Theory (DFT) and other electronic structure methods. Its primary role is to solve for the electron density of a molecular system by ensuring that the computed electronic potential and the resulting electron density are mutually consistent [1]. The SCF cycle involves repeatedly constructing the Fock matrix from the current density, diagonalizing it to obtain new orbitals, and building a new density matrix until the input and output densities converge within a specified threshold [2].
Achieving robust SCF convergence is a prerequisite for obtaining accurate predictions of molecular properties. The electron density directly determines all ground-state electronic properties, including molecular energies, reaction barriers, vibrational frequencies, and spectroscopic parameters [3] [1]. Poor convergence not only prevents calculations from completing but can lead to qualitatively incorrect results, such as convergence to high-energy excited states rather than the ground state, or significant errors in predicted molecular geometries and energies [3]. Within the context of energy grid parameter research for distribution system operators (DSOs), the challenges of SCF convergence find a conceptual parallel in achieving stable convergence in smart grid energy flow optimizations, where iterative solvers must balance multiple constraints to reach optimal operational states [4] [5].
The SCF Convergence Challenge
Fundamental Causes of Convergence Failure
SCF convergence failures predominantly arise from two scenarios: initial oscillations in the early iterations and trailing convergence where small, persistent changes prevent reaching the convergence threshold [3]. The former often occurs with poor initial guesses for the electron density, particularly for systems with complex electronic structures, such those involving transition metals, open-shell radicals, or near-degenerate orbital energy levels [2]. The latter represents a more insidious problem where the SCF cycle appears to progress but never formally converges, often due to numerical instabilities or the presence of multiple states with similar energies [3].
For ΔSCF calculations targeting excited states, the challenge intensifies, as the procedure requires converging to a saddle point on the electronic Hamiltonian rather than a minimum [3]. This necessitates specialized convergence methods to ensure the solution remains on the desired excited state surface, which is crucial for modeling processes like charge-transfer excitations and core-hole spectroscopies where time-dependent DFT (TDDFT) often fails [3].
Impact on Molecular Property Prediction
The accuracy of nearly all quantum chemically derived properties depends directly on the quality of the converged SCF solution. Forces used in geometry optimization are derived from the Hellmann-Feynman theorem, which requires a fully variational wavefunction only obtained at SCF convergence. Vibrational frequencies determined from the Hessian matrix (second derivatives of energy with respect to nuclear positions) are particularly sensitive to convergence quality, as small residual errors in the electron density can significantly affect curvature of the potential energy surface [3]. Properties like NMR chemical shifts, electronic circular dichroism (ECD), and vibrational circular dichroism (VCD) require highly accurate electron densities and orbital energies, yet open-source implementations for predicting these properties remain scarce [3].
Table 1: Molecular Properties and Their Dependence on SCF Convergence Quality
| Molecular Property | Dependence on SCF Convergence | Typical Convergence Requirement |
|---|---|---|
| Total Energy | Direct dependence on electron density accuracy | 10⁻⁶ a.u. (default in ADF) [2] |
| Nuclear Gradients (Forces) | Requires variational wavefunction for Hellmann-Feynman theorem | 10⁻⁶ a.u. or tighter |
| Molecular Geometry | Depends on accurate forces | 10⁻⁶ a.u. or tighter |
| Vibrational Frequencies | Highly sensitive to Hessian matrix accuracy | 10⁻⁸ a.u. or tighter [3] |
| Electronic Properties (HOMO/LUMO, Dipole Moment) | Direct dependence on orbital energies and electron density | 10⁻⁶ a.u. |
| Spectroscopic Parameters (NMR, ECD, VCD) | Requires highly precise density and orbital energies | 10⁻⁸ a.u. or tighter [3] |
Quantitative Assessment of SCF Convergence
Convergence Criteria and Metrics
The primary metric for SCF convergence is the commutator of the Fock and density matrices ([F,P]), which theoretically should be zero at full self-consistency [2]. In practical implementations, convergence is considered achieved when the maximum element of this commutator falls below a specified threshold (SCFcnv), while the norm of the matrix falls below 10×SCFcnv [2]. The ADF package implements a secondary criterion (sconv2) that, when met, allows calculations to continue with only a warning if the primary criterion cannot be achieved [2].
Table 2: Standard SCF Convergence Parameters in Quantum Chemistry Codes
| Parameter | Default Value in ADF | Function | Impact on Calculation |
|---|---|---|---|
| SCFcnv (Primary criterion) | 1.0×10⁻⁶ (Create mode: 1.0×10⁻⁸) | Threshold for maximum element of [F,P] commutator | Determines final convergence quality; tighter values needed for properties |
| sconv2 (Secondary criterion) | 1.0×10⁻³ | Fallback threshold when primary criterion not met | Allows continued computation with warning if moderate convergence achieved |
| Maximum Iterations (Niter) | 300 | Maximum SCF cycles before termination | Prevents infinite loops in problematic cases |
| DIIS N (Expansion vectors) | 10 | Number of previous cycles used in DIIS extrapolation | Critical for convergence acceleration; too small or large values can break convergence |
Performance of Acceleration Methods
Different SCF acceleration methods demonstrate varying performance characteristics across chemical systems. The mixed ADIIS+SDIIS method, used by default in ADF since 2016, typically provides optimal performance for most systems [2]. The LIST family of methods (LISTi, LISTb, LISTf) can be more effective for difficult cases but are sensitive to the number of expansion vectors [2]. The MESA method combines multiple acceleration techniques (ADIIS, fDIIS, LISTb, LISTf, LISTi, and SDIIS) and can be fine-tuned by disabling specific components for problematic systems [2].
Experimental Protocols for SCF Convergence
Standard Protocol for Routine Systems
For systems with well-behaved convergence characteristics, the following protocol provides reliable performance:
- Initialization: Use the default electron density guess (typically superposition of atomic densities or extended Hückel theory).
- SCF Parameters: Set convergence threshold to 1.0×10⁻⁶ a.u. for geometry optimizations and 1.0×10⁻⁸ a.u. for frequency calculations [3] [2].
- Acceleration: Employ the default ADIIS+SDIIS method with 10 DIIS expansion vectors [2].
- Monitoring: Track both the maximum element and norm of the [F,P] commutator to ensure proper convergence [2].
- Validation: Confirm convergence within the specified maximum iterations (typically 50-100 cycles for well-behaved systems).
Advanced Protocol for Problematic Systems
For systems exhibiting convergence difficulties (oscillatory behavior, trailing convergence, or failure to converge):
Enhanced Initial Guess:
- Utilize machine learning-based density prediction where available [3]
- Employ fragment-based or computational results file (CRF) restart from similar systems
Modified SCF Parameters:
- Implement the MESA method with selective component disabling (e.g.,
MESA NoSDIISfor oscillatory cases) [2] - Adjust DIIS expansion vectors (N=12-20) for LIST methods [2]
- For ADIIS, decrease thresholds (THRESH1 and THRESH2) to 0.001 and 0.00001 respectively to let A-DIIS approach the final solution [2]
- Implement the MESA method with selective component disabling (e.g.,
Alternative Strategies:
Diagram 1: SCF Convergence Workflow
Protocol for ΔSCF Excited State Calculations
For calculating excited states using the ΔSCF method:
- Initial Orbital Selection: Manually promote electrons to target the desired excited state configuration [3].
- Convergence Enforcement: Apply the Maximum Overlap Method (MOM) to maintain orbital character during iterations [3].
- Constrained DFT: Implement charge or spin constraints where appropriate for charge-transfer states [3].
- Validation: Confirm convergence to saddle point by verifying the Hessian has exactly one negative eigenvalue.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Software Tools for SCF Convergence Research
| Tool/Resource | Type | Primary Function | Application in SCF Research |
|---|---|---|---|
| ADF SCF Module [2] | Software Module | SCF convergence implementation | Provides production implementation of multiple acceleration methods |
| Libxc [3] | Software Library | Exchange-correlation functionals | Enables testing SCF convergence across functional types |
| Open Molecules 2025 [6] | Dataset | Training data for ML density guesses | Provides reference data for developing improved initial guesses |
| DeePMD-kit [1] | Software Framework | Neural network potentials | Alternative to DFT for large systems where SCF convergence is problematic |
| CREST [3] | Conformer Search | Metadynamics-based sampling | Generates diverse molecular geometries for testing SCF robustness |
| DP-GEN [7] | Active Learning | Automated potential generation | Framework for developing systems with guaranteed SCF convergence |
Advanced Applications and Future Directions
Machine Learning Approaches
Machine learning offers promising avenues for addressing SCF convergence challenges. ML-based electron density guesses can provide starting points much closer to the final solution, significantly reducing iteration counts [3]. Training such models requires large datasets of high-quality electron densities, such as the Open Molecules 2025 dataset with >100 million DFT calculations [6]. Transfer learning approaches, as demonstrated in the EMFF-2025 neural network potential, show that models pre-trained on diverse chemical systems can be specialized with minimal additional data [7].
Connection to Grid Parameter Optimization
The challenge of SCF convergence shares conceptual parallels with achieving convergence in smart grid energy management systems. Both involve iterative optimization of complex, nonlinear systems with multiple interacting components [5] [8]. Grid operators face analogous challenges in achieving convergence to stable operating points while managing distributed energy resources, where artificial neural networks (ANNs) and other AI methods are increasingly employed for optimization [5]. Research in either domain can inform the other, particularly in developing robust convergence accelerators and adaptive optimization parameters.
Diagram 2: SCF and Grid Convergence Analogy
Emerging Research Frontiers
Future research directions include developing universal SCF convergence accelerators that automatically adapt to system characteristics, eliminating the need for manual parameter tuning [3]. Hybrid quantum-classical algorithms may leverage quantum computers to calculate particularly challenging components of the SCF procedure. For drug development professionals, improved SCF convergence directly translates to more reliable prediction of protein-ligand binding energies, spectroscopic properties for characterization, and reaction mechanisms for synthetic planning [3]. The ongoing development of unified thermochemistry libraries and better implicit-solvent models will further increase the demands on SCF convergence for pharmaceutical applications [3].
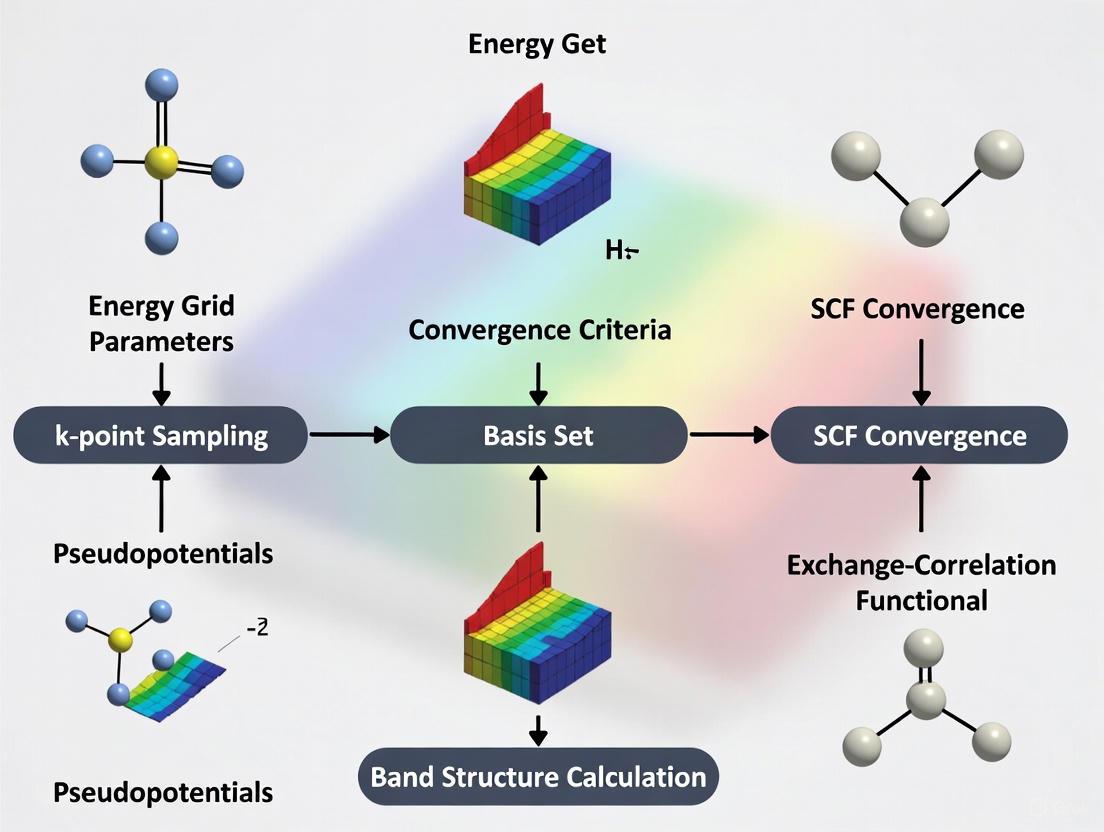
Self-Consistent Field (SCF) theory represents a cornerstone of modern computational quantum chemistry, enabling the in silico modeling of chemical reactions and the first-principles design of novel materials and catalysts [9]. As the simplest, most affordable, and most widely-used category of electronic structure methods, SCF approaches include both Hartree-Fock (HF) theory and Kohn-Sham density functional theory (DFT) [10]. The mathematical foundation of these methods lies in solving the SCF equations through an iterative procedure that continues until the energy is minimized and the electron distribution becomes consistent with the potential it generates [9]. This application note provides a comprehensive overview of the mathematical foundations of SCF theory, with particular emphasis on its relevance to convergence research, specifically in setting energy grid parameters for Density of States (DOS) convergence studies essential for drug development and materials science applications.
Theoretical Framework
Basis Set Expansion and the LCAO Ansatz
In the HF and DFT approaches, the electronic wave function is formulated as a Slater determinant where electrons occupy a set of molecular orbitals (MOs). Central to the SCF methodology is the Linear Combination of Atomic Orbitals (LCAO) ansatz, where MOs are expanded in terms of normalized atomic orbital basis functions [9]:
[ \varphii^\alpha(\vec{r}) = \sum{\mu=1}^M C{\mu i}^\alpha \chi\mu(\vec{r}) ]
[ \varphii^\beta(\vec{r}) = \sum{\mu=1}^M C{\mu i}^\beta \chi\mu(\vec{r}) ]
Here, ( \varphii^\alpha ) and ( \varphii^\beta ) represent the α (spin-up) and β (spin-down) molecular orbitals, ( C{\mu i}^\alpha ) and ( C{\mu i}^\beta ) are the expansion coefficients, and ( \chi\mu ) are the atomic orbital basis functions, with M indicating the total number of basis functions [9]. The basis functions are typically not orthonormal, with their overlap defined by the overlap matrix ( S{\mu\nu} ):
[ \int d\vec{r} \chi\mu(\vec{r}) \chi\nu(\vec{r}) = S{\mu\nu} \neq \delta{\mu\nu} ]
Table 1: Comparison of SCF Method Theoretical Foundations
| Method | Theoretical Basis | Electron Correlation Treatment | Computational Cost | Typical Applications |
|---|---|---|---|---|
| Hartree-Fock (HF) | Wavefunction theory | Mean-field, exact exchange | Moderate | Reference calculations, molecular properties |
| Density Functional Theory (DFT) | Electron density | Approximate exchange-correlation functional | Low to moderate | Large systems, catalysis, materials |
| Local Density Approximation (LDA) | Uniform electron gas model | Local density dependence | Low | Metallic systems, preliminary studies |
| Generalized Gradient Approximation (GGA) | Electron density and gradient | Semi-local functional | Low to moderate | General purpose, molecular systems |
| Meta-GGA | Density, gradient, and kinetic energy density | Higher-order semi-local | Moderate | Improved accuracy for diverse systems |
Density Matrices and Electron Density
The electron density plays a fundamental role in quantum chemistry, particularly in DFT calculations. The spin-σ electron density can be expressed as [9]:
[ \rho^\sigma(\vec{r}) = \sum{i=1}^{N\sigma} |\varphii^\sigma(\vec{r})|^2 = \sum{i=1}^{N\sigma} \sum{\mu\nu} C{\mu i}^\sigma C{\nu i}^\sigma \chi\mu(\vec{r}) \chi\nu(\vec{r}) = \sum{\mu\nu} P{\mu\nu}^\sigma \chi\mu(\vec{r}) \chi\nu(\vec{r}) ]
Here, ( N_\sigma ) represents the number of spin-σ electrons in the system, and the density matrix ( P^\sigma ) is defined as [9]:
[ P{\mu\nu}^\sigma = \sum{i=1}^{N\sigma} C{\mu i}^\sigma C_{\nu i}^\sigma ]
The total electron density is obtained from the sum of the α and β densities: ( \rho(\vec{r}) = \rho^\alpha(\vec{r}) + \rho^\beta(\vec{r}) ), with a corresponding total density matrix ( P = P^\alpha + P^\beta ) [9].
SCF Equations and Eigenvalue Problems
The SCF equations manifest as generalized eigenvalue problems in the non-orthogonal atomic orbital basis set. For restricted calculations, these take the form of the Roothaan-Hall equations [9]:
[ \mathbf{F} \mathbf{C} = \mathbf{S} \mathbf{C} \mathbf{E} ]
For unrestricted open-shell systems, the Pople-Nesbet-Berthier equations yield a coupled set of generalized eigenvalue equations [9]:
[ \mathbf{F}^\alpha \mathbf{C}^\alpha = \mathbf{S} \mathbf{C}^\alpha \mathbf{E}^\alpha ] [ \mathbf{F}^\beta \mathbf{C}^\beta = \mathbf{S} \mathbf{C}^\beta \mathbf{E}^\beta ]
In these equations, ( \mathbf{F} ) represents the Fock matrix, ( \mathbf{S} ) is the overlap matrix, ( \mathbf{C} ) contains the molecular orbital coefficients, and ( \mathbf{E} ) is a diagonal matrix of orbital energies [9].
SCF Convergence Protocols
Standard SCF Iterative Procedure
The solution of SCF equations typically employs an iterative procedure until self-consistency is achieved. The fundamental steps in this process are visualized in the following workflow:
Advanced Convergence Techniques
When standard SCF procedures fail to converge, several advanced strategies can be employed:
Initial Guess Modification: Changing the initial electron density guess is recommended when SCF calculations fail to converge, with options including superposition of atomic densities, fragment approaches, or results from previous calculations [10].
Convergence Algorithm Alteration: Switching between different convergence algorithms, such as damping, level shifting, or direct inversion in iterative subspace (DIIS) methods can stabilize convergence [10].
Dual-Basis Approaches: These methods facilitate large-basis quality results while requiring self-consistent iterations only in a smaller basis set, significantly improving computational efficiency [10].
SCF Meta-dynamics: This technique helps locate multiple solutions to the SCF equations and verifies that the obtained solution represents the lowest minimum [10].
Density of States Convergence Protocol
For DOS convergence research, particularly relevant for electronic structure analysis in drug development, the following detailed protocol is recommended:
Table 2: DOS Convergence Protocol Parameters
| Step | Parameter | Recommended Settings | Convergence Criterion | Remarks |
|---|---|---|---|---|
| Initialization | Basis Set | 6-31G* or def2-SVP | N/A | Balance between accuracy and cost |
| K-points Grid | 3×3×3 (minimal) | N/A | For periodic systems | |
| Energy Grid | 0.5 eV resolution | N/A | Initial coarse grid | |
| SCF Cycle | Max Iterations | 100-200 | Energy change < 10⁻⁶ Ha | Adjust based on system |
| Density Convergence | 10⁻⁶ a.u. | Density change < 10⁻⁵ | Critical for property accuracy | |
| Mixing Scheme | DIIS with 0.1 damping | Stable convergence | Reduce damping if oscillating | |
| DOS Refinement | K-point Grid | Increase to 6×6×6 | DOS features stable | Monitor band edges |
| Energy Grid | 0.05-0.01 eV | Peak positions stable | Focus on relevant energy window | |
| Broadening | 0.1-0.05 eV | Physical peak width | Gaussian/Lorentzian mixing |
Procedure:
System Preparation:
- Define molecular geometry or crystal structure
- Select appropriate basis set considering accuracy requirements and computational resources
- Choose exchange-correlation functional appropriate for the system (e.g., PBE for metals, B3LYP for molecules)
Initial SCF Calculation:
- Perform calculation with moderate convergence criteria (energy change < 10⁻⁵ Ha)
- Use coarse k-point grid for periodic systems (e.g., 3×3×3)
- Employ minimal energy grid resolution (0.5 eV) for initial DOS calculation
Convergence Assessment:
- Monitor total energy changes between iterations
- Track density matrix convergence
- Verify orbital energy stability, particularly for frontier orbitals
Grid Refinement:
- Systematically increase k-point density until DOS features stabilize
- Refine energy grid resolution to 0.05-0.01 eV in relevant energy regions
- Adjust broadening parameters to physically meaningful values
Validation:
- Compare DOS with experimental data when available
- Verify integration of DOS gives correct electron count
- Check consistency of band gaps or HOMO-LUMO gaps with expected values
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Tool/Component | Function/Purpose | Implementation Examples | Relevance to DOS Convergence |
|---|---|---|---|
| Basis Sets | Atomic orbital basis for MO expansion | Gaussian-type orbitals (GTOs), Slater-type orbitals (STOs), numerical AOs (NAOs) | Determines accuracy of wavefunction representation and computational cost |
| Exchange-Correlation Functionals | Approximate electron correlation in DFT | LDA, GGA (PBE), meta-GGA (SCAN), hybrid (B3LYP) | Critical for accurate electronic structure and DOS features |
| K-point Grids | Brillouin zone sampling for periodic systems | Monkhorst-Pack scheme, Gamma-centered | Essential for convergent DOS in materials and surfaces |
| Density Matrix | Electron density representation in basis | Construction from MO coefficients | Directly determines accuracy of calculated electron density |
| SCF Convergers | Algorithms for SCF convergence | DIIS, EDIIS, damping, level shifting | Enable stable convergence to ground state for accurate DOS |
| Energy Grid Parameters | DOS energy point discretization | Resolution, energy range, broadening | Controls resolution and smoothness of final DOS spectrum |
| Pseudopotentials/ECPs | Core electron approximation | Norm-conserving, ultrasoft, PAW | Reduces computational cost while maintaining valence electron accuracy |
Convergence Framework and Methodology
The convergence of SCF calculations, particularly in the context of DOS analysis for materials and drug development applications, requires a systematic approach. Recent research emphasizes convergence as problem-driven research that fosters deep integration across disciplines [11]. This is especially relevant when setting energy grid parameters for DOS convergence, where both technical parameters and physical understanding must be integrated.
The mathematical framework for assessing convergence involves monitoring multiple parameters:
Energy Convergence: The total energy difference between successive iterations should approach zero: [ \Delta E = |E{n} - E{n-1}| < \epsilon_E ]
Density Convergence: The change in the density matrix should diminish: [ \Delta P = ||P{n} - P{n-1}|| < \epsilon_P ]
DOS Convergence: The density of states should become invariant to further k-point or energy grid refinement: [ \text{DOS}(E, \text{grid}{n}) - \text{DOS}(E, \text{grid}{n-1}) \approx 0 ]
The relationship between these convergence criteria and the computational workflow can be visualized as follows:
For research focusing on drug development applications, particular attention should be paid to the accurate calculation of frontier orbital energies (HOMO-LUMO gap) and the DOS in the energy region relevant to molecular interactions, as these parameters directly influence binding affinity and reactivity predictions.
The mathematical foundations of Self-Consistent Field theory provide the essential framework for electronic structure calculations central to modern computational chemistry and materials science. The LCAO approach, combined with efficient iterative diagonalization techniques, enables the solution of the SCF equations for both molecular and periodic systems. For Density of States convergence research, careful attention to basis set selection, k-point sampling, energy grid parameters, and convergence criteria is essential for obtaining reliable results. The protocols and methodologies outlined in this application note provide researchers with a systematic approach to setting appropriate energy grid parameters for DOS convergence, facilitating accurate electronic structure calculations in drug development and materials design applications. As SCF methodologies continue to evolve, particularly with advances in linear-scaling algorithms and improved density functional approximations, the efficiency and applicability of these methods to larger and more complex systems will further expand their utility in scientific research and industrial applications.
Common Convergence Challenges in Biomolecular Systems and Transition Metal Complexes
A critical, yet often overlooked, step in computational chemistry is ensuring the convergence of the electron density of states (DOS) and related properties. This process involves defining appropriate energy grid parameters and computational settings to achieve a stable, accurate numerical solution of the electronic structure. Inefficient or incomplete convergence can lead to inaccurate energies, forces, reaction barriers, and spectroscopic predictions, fundamentally compromising the reliability of a simulation. These challenges are particularly acute in two important classes of systems: large, flexible biomolecules and electronically complex transition metal complexes (TMCs). This application note details the specific convergence challenges encountered in these systems and provides validated protocols to overcome them.
The table below summarizes the core sources of convergence difficulties for biomolecular systems and transition metal complexes, highlighting the distinct nature of the problems in each domain.
Table 1: Fundamental Convergence Challenges in Biomolecular and Transition Metal Systems
| System Characteristic | Biomolecular Systems (e.g., Proteins, DNA, in Solvent) | Transition Metal Complexes (TMCs) |
|---|---|---|
| Primary Challenge | System size and conformational flexibility [12] | Strong electron correlation and multi-configurational ground states [13] |
| Typical System Size | 2 to 350+ atoms per snapshot [12] | Varies, but often smaller (e.g., 10-100 atoms) |
| Key Electronic Structure Problem | Accurate treatment of diverse non-covalent interactions (electrostatics, dispersion) [12] | Description of near-degenerate d-orbitals, metal-ligand charge transfer, and spin states [13] |
| Impact on DOS/Energy Convergence | Slow convergence with basis set size; sensitivity to functional for dispersion; requires large integration grids [12] | High sensitivity to the choice of density functional approximation (DFA); instability in SCF cycles due to near-degeneracies [13] |
| Recommended Functional Class | Range-separated meta-GGAs (e.g., ωB97M-V) [12] | Multiple DFAs across Jacob's Ladder; consensus approach recommended [13] |
Protocols for DOS Convergence
Protocol for Biomolecular Systems
This protocol is optimized for achieving DOS and energy convergence in large biomolecular systems, including proteins, nucleic acids, and their complexes with ligands in explicit solvent.
1. System Preparation and Pre-Optimization
- Structure Source: Obtain initial coordinates from experimental databases (e.g., RCSB PDB) or generate using tools like Architector [12].
- Protonation and Tautomers: Use tools like Schrödinger to sample biologically relevant protonation states and tautomers [12].
- Solvation: Employ explicit solvent models (e.g., TIP3P) within a QM/MM framework where the core region is treated quantum mechanically and the environment with a molecular mechanical force field [14].
2. Electronic Structure Method Selection
- Density Functional: Use the ωB97M-V functional with the def2-TZVPD basis set. This meta-GGA functional provides a high-accuracy, balanced description of various interaction types prevalent in biomolecules [12].
- Integration Grid: Select a large, pruned grid (e.g., 99,590 points) to ensure accurate integration, which is critical for gradients and non-covalent interactions [12].
3. Self-Consistent Field (SCF) Convergence
- Algorithm: Use the Gaussian Smearing method with an initial smearing width of 0.01–0.05 eV to aid initial convergence by populating near-degenerate states, then reduce the width for the final calculation.
- Mixing Parameters: For difficult systems, increase the SCF density mixing parameter (e.g., to 0.05–0.10) or use a Kerker model to damp long-range charge oscillations.
- Fallback: If SCF fails, use the "Always Generate Initial Guess" option to recalculate the initial density from core Hamiltonians.
4. DOS Calculation and Analysis
- Energy Grid: For post-processing DOS, set a fine energy grid with a k-point spacing of ≤ 0.01 eV/Angstrom if using periodic boundary conditions. For molecular clusters, a high density of states points (e.g., 1000 points/eV) is recommended.
- Broadening: Apply a small Gaussian broadening (e.g., 0.01-0.05 eV) to the calculated DOS to smooth the distribution and aid in visualization and analysis, ensuring this does not obscure genuine physical features.
The following workflow diagram outlines the key steps and decision points in this protocol.
Protocol for Transition Metal Complexes
This protocol addresses the severe convergence challenges in TMCs, which stem from their complex electronic structure, including multi-reference character and narrow energy gaps.
1. Active Learning and System Sampling
- Design Space Construction: Build a space of synthetically accessible TMCs using ligand databases (e.g., Cambridge Structural Database). For chromophores, constrain to octahedral d⁶ Fe(II) or Co(III) centers with bidentate ligands [13].
- Consensus DFT Approach: To mitigate DFA bias, employ an ensemble of 23 density functionals across multiple rungs of Jacob's Ladder for property evaluation. This identifies candidates where predictions are robust [13].
2. Electronic Structure Method Selection
- Functional Selection: No single functional is universally best. Use a multi-functional consensus or select a hybrid functional known for reasonable performance on TMCs (e.g., a member of the Minnesota family). Always test sensitivity.
- Basis Set: Use a triple-zeta quality basis set with polarization functions (e.g., def2-TZVP). For metals, incorporate diffuse functions if studying excited states or anion properties.
- Integration Grid: Use an ultrafine grid (e.g., 150,000 points or more) to accurately capture the complex electron density around the metal center.
3. Advanced SCF Convergence
- Initial Guess: Use a fragment-based or atomic guess rather than a superposition of atomic densities (SAD) to provide a better starting point for the metal-ligand system.
- Smearing and Damping: Apply a larger initial smearing width (0.05–0.10 eV) than for biomolecules. Combine this with robust damping algorithms (e.g., a combination of Kerker and Thomas-Fermi screening).
- Stability Analysis: After initial convergence, perform a wavefunction stability check. If an unstable solution is found, re-optimize using the unstable wavefunction as a new guess.
4. DOS and Property Validation
- Multi-Reference Character: Calculate the %t1 diagnostic or the ℏND parameter from fractional occupation number DFT. A value of ℏND > 0.307 indicates strong multi-reference character, signaling that single-reference DFT may be inadequate [13].
- Target Properties: For chromophores, calculate the Δ-SCF absorption energy and ensure it falls within the target visible range (1.5–3.5 eV). Verify the ground state is low-spin to promote desired metal-to-ligand charge-transfer states [13].
- DOS Analysis: Analyze the projected DOS (PDOS) onto the metal d-orbitals and ligand orbitals to confirm the expected electronic structure and identify the HOMO-LUMO gap character.
The workflow for TMCs involves careful state preparation and validation, as shown below.
The Scientist's Toolkit: Essential Research Reagents and Computational Solutions
The following table details key computational tools and datasets essential for conducting the research described in this application note.
Table 2: Key Research Reagent Solutions for Convergence Studies
| Tool/Resource Name | Type | Primary Function in Research | Application Context |
|---|---|---|---|
| Open Molecules 2025 (OMol25) [12] | Dataset | Provides over 100 million gold-standard DFT calculations for training and benchmarking machine learning interatomic potentials and validating methods. | Biomolecules, Electrolytes, Metal Complexes |
| Universal Model for Atoms (UMA) [12] | Pre-trained Model | A universal neural network potential trained on OMol25 and other datasets for fast, accurate energy and force predictions. | All System Types |
| Architector Package [12] | Software Tool | Generates initial 3D geometries for metal complexes combinatorially using GFN2-xTB, providing starting structures for high-level calculation. | Transition Metal Complexes |
| ωB97M-V/def2-TZVPD [12] | Computational Method | A high-accuracy density functional and basis set combination used for generating reference data in the OMol25 dataset. | All System Types |
| MiMiC Framework [14] | Simulation Framework | Enables highly efficient multi-scale QM/MM MD simulations by coupling different computational chemistry programs optimally. | Biomolecular Systems |
| rND (nondynamical correlation metric) [13] | Diagnostic Metric | Quantifies multireference character from fractional occupation number DFT; values > ~0.3 indicate potential single-reference DFT failure. | Transition Metal Complexes |
| Active Learning with 2D Efficient Global Optimization [13] | Computational Workflow | Balances exploration and exploitation to efficiently discover target molecules (e.g., chromophores) from vast chemical spaces. | Transition Metal Complexes |
Impact of Convergence Failures on Drug Discovery Timelines and Resource Allocation
Convergence failures, the breakdown in integrating data and methodologies across disciplines, represent a critical bottleneck in modern drug discovery. The traditional linear, siloed approach to research and development (R&D) contributes significantly to the sector's well-documented productivity crisis, characterized by unsustainable costs and extended timelines. Eroom's Law—the observation that the number of new drugs approved per billion US dollars spent has halved roughly every nine years since 1950—illustrates this worsening inefficiency [15]. This application note quantifies the impact of convergence failures on development timelines and resource allocation, provides validated experimental protocols to diagnose and remediate such failures, and establishes a framework for optimizing energy grid parameters to ensure robust Density of States (DOS) convergence in computational drug discovery.
Quantitative Impact of Convergence Failures
The failure to effectively integrate data from patents, scientific literature, clinical trials, and real-world evidence creates significant downstream inefficiencies and costs. The following tables summarize the quantitative impact on timelines, costs, and success rates.
Table 1: Impact of Traditional Silos vs. Convergent Approaches on Development Metrics
| Development Metric | Traditional Siloed Approach | Integrated Convergent Approach | Impact of Convergence |
|---|---|---|---|
| Average Timeline | 10–15 years [16] [15] | 5–7.5 years (Projected 50% reduction) [17] | 50% reduction |
| Average Cost per Approved Drug | $2.6 billion [15] | Significant reduction via early failure [17] [18] | Avoids costly late-stage failures |
| Clinical Trial Success Rate | ~10% (90% failure rate) [19] [15] | Increased via better target validation & patient stratification [18] | Potential for substantial improvement |
| Probability of Phase I to Approval | 13.8% overall (range 3.4–33.4%) [20] | Higher predicted success with integrated data [21] | Mitigates Phase II "graveyard" |
Table 2: Phase-by-Phase Attrition and Primary Causes of Failure
| Development Phase | Attrition Rate | Primary Causes of Failure (Often due to Convergence Gaps) |
|---|---|---|
| Preclinical | High (>99% of candidates fail) [16] | Poor target validation, unforeseen toxicity in animal models [20] [15] |
| Phase I | ~37% [15] | Safety and dosage issues in humans |
| Phase II | ~70% [15] | Lack of efficacy in patients—major convergence failure point |
| Phase III | ~42% [15] | Insufficient efficacy vs. standard of care, safety in larger population |
Experimental Protocols for Diagnosing Convergence Failures
Protocol: Integrated Data Audit for Target Validation
Purpose: To systematically identify gaps and inconsistencies in the biological, chemical, and clinical data supporting a proposed drug target before initiating costly screening campaigns.
Materials:
- Life Science Knowledge Graph (e.g., BenevolentAI Platform): Integrates public and proprietary data from scientific literature, patents, genomics, and clinical trials [22] [23].
- CETSA (Cellular Thermal Shift Assay) Kit: Validates direct target engagement of a drug molecule within intact cells, providing physiologically relevant confirmation [24].
- hiPSC-derived Disease Models: Human induced pluripotent stem cell-derived cells (e.g., neurons, microglia) for pathophysiologically relevant testing in complex diseases [20].
- AI-Powered Target Prediction Software: Uses machine learning to analyze genomic, proteomic, and patient data to identify and prioritize novel disease targets [15] [18].
Procedure:
- Data Mapping: Using the knowledge graph, map all known interactions, pathways, and genetic associations for the proposed target. Flag any inconsistencies between data types (e.g., genomic data suggests efficacy, but proteomic data does not) [23].
- Competitive Landscape Analysis: Audit patent and clinical trial databases to assess competitor activity and identify potential overlapping mechanisms or prior art that might limit the freedom to operate [23].
- Experimental Validation: In the hiPSC-derived disease model, perform CETSA assays to confirm that a known modulator of the target engages and stabilizes it in a dose-dependent manner [24] [20].
- Go/No-Go Decision: Integrate the computational and experimental findings. A "Go" decision requires consistent supporting evidence across all data pillars and confirmation of direct target engagement in a physiologically relevant human model.
Protocol: Quantum Chemical Calculation with DOS Convergence Monitoring
Purpose: To ensure robust and reproducible electronic structure calculations for in silico drug design by achieving DOS convergence, thereby preventing wasted computational resources and erroneous predictions.
Materials:
- High-Performance Computing (HPC) Cluster: Configured with quantum chemistry software (e.g., VASP, Gaussian).
- Molecular Dataset: A curated set of small molecule drug candidates with known electronic properties for validation.
Procedure:
- Initial Setup: Define the molecular system and select an appropriate basis set and exchange-correlation functional.
- Parameter Sweep: Systematically vary the energy grid cut-off (ECUT) and K-point mesh density parameters. These directly control the resolution and sampling of the energy space, critical for DOS convergence.
- Calculation Execution: For each parameter set, run a single-point energy calculation and extract the total energy and DOS.
- Convergence Criteria Check: Plot the total energy and the highest occupied molecular orbital (HOMO) – lowest unoccupied molecular orbital (LUMO) gap against the ECUT and K-point parameters. Convergence is achieved when these values change by less than a predefined threshold (e.g., 1 meV/atom) between successive parameter refinements.
- Validation: Once the optimal parameters are identified, run calculations on the validation dataset to ensure predicted properties (e.g., ionization potential, electron affinity) align with reference data.
Diagram 1: DOS convergence workflow.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Materials and Platforms for Integrated Discovery
| Research Reagent / Platform | Function & Application | Role in Preventing Convergence Failure |
|---|---|---|
| CETSA Assay Kits [24] | Measures drug-target engagement in intact cells and tissues under physiological conditions. | Bridges the gap between biochemical potency and cellular efficacy, a key point of failure. |
| hiPSC-Derived Cells & Organoids [20] | Provides human-specific, pathologically relevant models for efficacy and toxicity testing. | Reduces reliance on animal models, which have poor external validity for human responses. |
| Organs-on-Chips [20] | Microfluidic devices that recapitulate human organ-level physiology and tissue-tissue interfaces. | Enables more accurate human PK/PD modeling and assessment of complex drug effects. |
| AI-Driven Knowledge Graphs (e.g., BenevolentAI, Exscientia) [22] [23] | Integrates disparate data sources (patents, literature, omics, trials) to identify novel targets and connections. | Systematically identifies inconsistencies and gaps in the early hypothesis, forcing convergence. |
| Generative AI Chemistry Platforms (e.g., Insilico Medicine) [22] [15] | Designs novel molecular structures from scratch optimized for multiple properties (potency, ADMET). | Compresses design cycles and generates molecules optimized for both efficacy and developability. |
| Federated Data Platforms (e.g., Lifebit) [15] | Enables secure, compliant analytics across distributed clinical and genomic datasets without moving data. | Allows integration of real-world evidence into discovery while maintaining privacy, improving translational predictivity. |
Visualizing the Convergent Workflow
The following diagram illustrates an integrated, AI-driven drug discovery workflow designed to systematically prevent convergence failures by establishing continuous feedback loops between computational and experimental data.
Diagram 2: Convergent AI-driven discovery workflow.
Practical Implementation: Method Selection and Parameter Configuration Strategies for Stable Convergence
Self-Consistent Field (SCF) methods are fundamental computational procedures in quantum chemistry for determining molecular electronic structure. This application note provides a detailed comparative analysis of three prominent SCF convergence algorithms—DIIS, MultiSecant, and LIST methods—within the specific research context of setting energy grid parameters for Density of States (DOS) convergence. We present structured performance comparisons, detailed experimental protocols, and visualization tools to guide researchers in selecting and implementing optimal SCF methodologies for electronic structure calculations in materials science and drug development applications.
The Self-Consistent Field (SCF) procedure is an iterative computational method at the heart of modern quantum chemical calculations, particularly in Density Functional Theory (DFT). In Kohn-Sham DFT, the total electronic energy is expressed as a functional of the electron density, and the SCF method searches for a self-consistent electron density where the input and output densities converge [25]. The convergence is typically monitored through the self-consistent error, defined as the square root of the integral of the squared difference between the input and output density: (\text{err}=\sqrt{\int dx \; (\rho\text{out}(x)-\rho\text{in}(x))^2 }) [26].
Achieving SCF convergence presents significant computational challenges, particularly for systems with complex electronic structures such as transition metal complexes, open-shell systems, and molecules with small HOMO-LUMO gaps. The choice of convergence algorithm directly impacts computational efficiency, stability, and reliability of results—factors critically important for DOS calculations where accurate convergence directly influences electronic property predictions [27]. This application note focuses on three principal algorithms—DIIS, MultiSecant, and LIST methods—providing researchers with practical implementation guidelines within the context of electronic structure calculations for materials and pharmaceutical development.
Comparative Algorithm Analysis
Algorithm Specifications and Mechanisms
Direct Inversion in the Iterative Subspace (DIIS) is one of the most widely used SCF convergence algorithms. The DIIS method accelerates convergence by constructing an optimal linear combination of previous trial density matrices or Fock matrices to generate an improved guess for the next iteration [26] [28]. This extrapolation procedure effectively reduces oscillations in the convergence path. Key parameters controlling DIIS performance include the number of previous vectors retained in the expansion (NVctrx), damping parameters (DiMix, DiMixMin, DiMixMax), and criteria for handling large expansion coefficients (CHuge, CLarge) [26].
MultiSecant Methods represent a class of quasi-Newton approaches that generalize the secant method to multidimensional problems. These methods build an approximate Jacobian matrix using information from previous iterations, effectively capturing the convergence landscape without explicit Jacobian calculation [26]. In the SCM software implementation, MultiSecant serves as an alternative to DIIS at similar computational cost per cycle and can offer improved convergence in problematic cases [26]. The method is particularly valuable for systems where DIIS exhibits oscillatory behavior.
LIST Methods (including LISTi, LISTb, and LISTd variants) constitute a family of algorithms implemented as alternatives within the DIIS framework [26]. These methods employ different strategies for handling the iterative subspace and managing the history of iterations. The LIST variants provide flexibility in managing the balance between convergence stability and computational overhead, with each variant employing distinct approaches to building and maintaining the iterative subspace.
Performance Comparison Table
Table 1: Comparative Characteristics of SCF Convergence Algorithms
| Algorithm | Computational Efficiency | Convergence Stability | Memory Requirements | Optimal Application Domain | Key Tunable Parameters |
|---|---|---|---|---|---|
| DIIS | High for standard systems | Moderate; prone to oscillations in difficult cases | Moderate (stores 5-10 previous vectors) | Standard molecular systems with reasonable HOMO-LUMO gaps | NVctrx, DiMix, CHuge, CLarge, Condition [26] |
| MultiSecant | Comparable to DIIS per cycle | High; robust for problematic convergence | Similar to DIIS | Systems with difficult convergence, metallic systems, small-gap semiconductors | Mixing parameters, history length [26] |
| LIST Methods | Variable by variant | Generally high with proper variant selection | Similar to DIIS | Systems requiring specialized subspace handling | Variant selection (LISTi, LISTb, LISTd), subspace management [26] |
| MultiStepper | Flexible, adaptive | High through preset pathways | Implementation-dependent | General purpose, black-box applications | Preset path configurations [26] |
Parameter Optimization Guidelines
Table 2: Key Algorithm Parameters and Optimization Recommendations
| Parameter | Algorithm | Default Value | Optimization Guidance | DOS Convergence Consideration |
|---|---|---|---|---|
Iterations |
All | 300 (SCM) [26], 50 (Q-Chem) [28] | Increase to 500+ for difficult systems | Essential for metallic systems with dense DOS near Fermi level |
Mixing |
All | 0.075 [26] | Reduce for oscillating systems; increase for monotonic convergence | Critical for DOS accuracy; affects orbital energy convergence |
Criterion |
All | Depends on NumericalQuality and ( \sqrt{N_\text{atoms}} ) [26] |
Tighten to 1e-7 or 1e-8 for property calculations | Directly impacts DOS resolution; tighter criteria needed for accurate band edges |
NVctrx |
DIIS | Implementation-dependent | 6-10 for standard systems; reduce if unstable | Affects convergence stability for systems with complex DOS features |
DiMix |
DIIS | Implementation-dependent | Adaptive mixing often preferable | Influences convergence rate of valence and conduction band states |
ElectronicTemperature |
All | 0.0 [26] | 500-5000 K for metallic systems | Essential for smearing DOS near Fermi level in metallic systems |
Experimental Protocols for SCF Convergence
Standard Convergence Protocol for DOS Calculations
This protocol provides a systematic approach for achieving SCF convergence in Density of States calculations, particularly relevant for systems with challenging electronic structures.
Initialization Phase:
- System Preparation: Begin with a reasonable initial geometry. For DOS calculations, ensure the k-point grid is sufficiently dense to capture electronic structure features (typically 0.02 Å⁻¹ or finer spacing in reciprocal space).
- Initial Density Guess: Select appropriate initial density strategy (
InitialDensityparameter). Use atomic orbital superposition (psi) for molecular systems; considerfrompotfor periodic systems or metallic clusters [26]. - Baseline Parameters: Set initial SCF parameters to conservative values:
Mixing = 0.05,Iterations = 200, and standard convergence criteria (Criterion = 1e-5 √N_atomsforNumericalQuality = Normal) [26].
SCF Execution Phase:
- Algorithm Selection: Begin with DIIS algorithm for standard systems. For metallic systems or those with small HOMO-LUMO gaps (<0.1 eV), consider MultiSecant or LIST methods as primary alternatives.
- Convergence Monitoring: Track both energy change and density change between iterations. For DOS calculations, also monitor the stability of frontier orbital energies (HOMO and LUMO).
- Adaptive Adjustment: If oscillations occur after 20-30 iterations, reduce mixing parameter by 30-50% or switch to MultiSecant method. If convergence stagnates, gradually increase mixing parameter or consider LIST variant methods.
Convergence Validation:
- Accuracy Verification: Confirm that final SCF error is at least one order of magnitude smaller than the desired DOS energy resolution.
- DOS Calculation: Proceed with DOS calculation only after stable SCF convergence is achieved, verifying that electronic temperature settings (if used) are appropriate for the system [26].
Troubleshooting Protocol for Problematic Systems
For systems exhibiting persistent SCF convergence failures, implement this structured troubleshooting approach:
Diagnosis Phase:
- Convergence Pattern Analysis: Examine the SCF energy progression. Oscillatory behavior suggests reducing mixing parameters; monotonic but slow convergence benefits from increased mixing or algorithm switching [27].
- Electronic Structure Analysis: Check HOMO-LUMO gap at final geometry. Gaps smaller than kT suggest implementing fractional occupancy smearing (
ElectronicTemperature= 500-5000 K) [26]. - Wavefunction Stability: Perform initial stability analysis using
ROBUST_STABLEalgorithm if available [28] to ensure ground state convergence.
Intervention Phase:
- Accuracy Enhancement: Increase integration grid density (
NumericalQuality = GoodorVeryGood), tighten SCF convergence criterion to 1e-7 or higher, and consider exact exchange-correlation potential evaluation [27]. - Algorithm Cycling: Implement sequential algorithm strategy: Begin with DIIS for initial convergence, switch to MultiSecant or LIST methods if stagnation occurs near convergence.
- Degeneracy Handling: For systems with near-degenerate states, enable
Degeneratekeyword with appropriate energy width (default 1e-4 a.u.) to smooth orbital occupations [26].
Advanced Strategies:
- Damping Techniques: Implement aggressive damping (mixing = 0.01-0.02) for initial 10-15 iterations, gradually increasing to standard values.
- Fallback Protocol: If standard methods fail, employ specialized algorithms:
RCA_DIISorADIIS_DIISfor initial convergence establishment, switching toDIIS_GDMfor final convergence [28].
Table 3: Essential Research Reagent Solutions for SCF Convergence Studies
| Resource | Function in SCF Convergence | Implementation Examples | Application Context |
|---|---|---|---|
| Integration Grids | Numerical integration of XC functional | UltraFine grid (Gaussian) [29], Various grid levels (PySCF) [30] | Critical for accuracy; denser grids needed for difficult convergence |
| Basis Sets | Represent molecular orbitals | TZ2P, 6-31G(d), cc-pVDZ [27] [31] | Larger bases need tighter convergence criteria |
| XC Functionals | Define exchange-correlation energy | B3LYP, PBE, wB97XD [29] [31] | Hybrid functionals often need tighter convergence than GGAs |
| Relativistic Methods | Account for relativistic effects | ZORA, Pauli formalism [27] | Essential for heavy elements; ZORA preferred over Pauli |
| Solvation Models | Incorporate solvent effects | SCRF, COSMO, SMD | Implicit solvation can improve or hinder convergence |
| Dispersion Corrections | Account for van der Waals interactions | D2, D3, VV10 [29] | Can affect convergence stability in dense systems |
Visualization of SCF Algorithm Workflows
SCF Convergence Algorithm Decision Workflow
DOS Convergence Optimization Pathway
The selection and optimization of SCF convergence algorithms—DIIS, MultiSecant, and LIST methods—represent a critical step in ensuring accurate and efficient electronic structure calculations, particularly for Density of States determinations in materials research and drug development. DIIS offers robust performance for standard systems, MultiSecant provides enhanced stability for challenging metallic or small-gap systems, while LIST methods deliver specialized subspace handling capabilities. Through the implementation of the structured protocols, parameter optimization strategies, and diagnostic workflows presented in this application note, researchers can systematically address SCF convergence challenges within the specific context of DOS calculations. The integrated approach of algorithm selection, parameter tuning, and systematic troubleshooting enables reliable convergence across diverse chemical systems, forming a foundation for accurate electronic property predictions in both materials design and pharmaceutical development.
Optimal Mixing Parameter (SCF%Mixing) Configuration for Different Molecular Systems
The self-consistent field (SCF) method is the fundamental algorithm for solving the electronic structure problem in density functional theory (DFT) and Hartree-Fock calculations [32]. This iterative procedure requires the electron density or Hamiltonian to converge to a stable solution, but this process can be challenging, slow, or even divergent without proper parameterization [33] [32]. The choice of optimal mixing parameters—which control how the new density or Hamiltonian is constructed from previous iterations—varies significantly across different molecular systems and is crucial for computational efficiency and accuracy [33] [34].
For researchers focusing on density of states (DOS) convergence, which requires particularly high accuracy in electronic structure calculations [35], appropriate SCF mixing strategy is even more critical. This application note provides structured guidelines and experimental protocols for determining optimal SCF mixing parameters across diverse molecular systems, with special consideration for DOS-related research.
Theoretical Background: SCF Convergence and Mixing Strategies
The SCF Cycle and Convergence Monitoring
The SCF cycle represents an iterative loop where the Kohn-Sham equations must be solved self-consistently: the Hamiltonian depends on the electron density, which in turn is obtained from the Hamiltonian [33]. Starting from an initial guess, the code computes the Hamiltonian, solves the Kohn-Sham equations to obtain a new density matrix, and repeats until convergence is reached [33].
Convergence is typically monitored through two main criteria:
- Density Matrix Tolerance: The maximum absolute difference (dDmax) between matrix elements of new and old density matrices, controlled by
SCF.DM.Tolerance(default: 10⁻⁴) [33] [34] - Hamiltonian Tolerance: The maximum absolute difference (dHmax) between matrix elements of the Hamiltonian, controlled by
SCF.H.Tolerance(default: 10⁻³ eV) [33] [34]
Both criteria must be satisfied by default for convergence, though either can be disabled if necessary [34].
Mixing Methods and Algorithms
SCF convergence relies heavily on the strategy for mixing the electron density or Hamiltonian between iterations. The two fundamental approaches are:
- Density Mixing: The density matrix (DM) is mixed between iterations [33] [34]
- Hamiltonian Mixing: The Hamiltonian (H) is mixed between iterations (typically provides better results) [33] [34]
Within these approaches, several algorithmic implementations exist:
- Linear Mixing: Uses a simple damping factor (
SCF.Mixer.Weight); robust but inefficient for difficult systems [33] - Pulay Mixing: Also known as Direct Inversion in the Iterative Subspace (DIIS); the default in many codes [33]
- Broyden Mixing: A quasi-Newton scheme that sometimes outperforms Pulay for metallic or magnetic systems [33]
The more sophisticated Pulay and Broyden methods retain a history of previous DMs or Hamiltonians (controlled by SCF.Mixer.History) to accelerate convergence [33] [34].
Optimal Mixing Parameters for Different System Types
Parameter Recommendations by System Category
Table 1: Optimal SCF Mixing Parameters for Different Molecular Systems
| System Type | Recommended Mixing Method | Optimal Weight | History Length | Special Considerations |
|---|---|---|---|---|
| Small Molecules (e.g., CH₄) | Pulay or Broyden [33] | 0.1-0.5 [33] | 2-4 [33] | Relatively easy to converge; default parameters often sufficient |
| Metallic Systems | Broyden [33] [36] | 0.1-0.3 [36] | 4-8 [36] | Requires smaller weights for stability; electron smearing recommended [32] [36] |
| Magnetic Systems (e.g., Fe clusters) | Broyden [33] | 0.1-0.3 (charge), 0.8 (spin) [36] | 4-8 [33] | For non-collinear calculations with difficult convergence, set mixing_angle=1.0 [36] |
| Open-Shell Systems | Pulay or Broyden [32] | 0.1-0.3 [32] | 4-8 [32] | Ensure correct spin multiplicity; strongly fluctuating errors may indicate improper electronic structure description [32] |
| Systems with Small HOMO-LUMO Gap | Broyden with electron smearing [32] | 0.05-0.2 [32] | 6-10 [32] | Fractional occupation numbers help overcome convergence issues [32] |
| Challenging/Divergent Systems | Linear or Pulay with reduced weight [32] | 0.015-0.09 [32] | 15-25 [32] | Use DIIS N=25, Cyc=30, Mixing=0.015, Mixing1=0.09 for slow but stable convergence [32] |
Advanced Mixing Techniques for Difficult Systems
For particularly challenging systems, additional techniques beyond standard mixing approaches may be necessary:
Electron Smearing: Applies fractional occupation numbers to distribute electrons over near-degenerate levels; particularly helpful for metallic systems or those with small HOMO-LUMO gaps [32] [36]. Keep the smearing value as low as possible and use successive restarts with reduced values [32].
Level Shifting: Artificially raises the energy of unoccupied orbitals; helpful for convergence but invalidates properties involving virtual orbitals (excitation energies, response properties, NMR shifts) [32].
U-Ramping for DFT+U: For systems using the DFT+U method, employ U-ramping with
mixing_restart>0andmixing_dmr=1to improve convergence [36].MESA Method: The MESA method combines multiple acceleration techniques (ADIIS, fDIIS, LISTb, LISTf, LISTi, and SDIIS) and can be particularly effective for problematic cases [2] [32].
Experimental Protocols for Parameter Optimization
General Workflow for Determining Optimal Mixing Parameters
The following diagram illustrates the systematic workflow for determining optimal SCF mixing parameters:
SCF Parameter Optimization Workflow
Protocol 1: Basic Mixing Parameter Screening
Purpose: To efficiently identify promising mixing parameter ranges for a new molecular system.
Materials and Setup:
- Quantum chemistry code (SIESTA, ADF, ABACUS, or VASP)
- Molecular structure file
- Baseline computational parameters (functional, basis set, k-points)
Procedure:
- Begin with a moderate number of maximum SCF iterations (e.g., 100) [33]
- Set mixing method to Pulay/DIIS (default in most codes) [33]
- Test a range of mixing weights (0.1, 0.2, 0.3, 0.5, 0.7) with default history length [33]
- For each parameter set, record:
- Number of iterations to convergence
- Final energy
- Convergence behavior (smooth, oscillatory, divergent)
- Computational time
- Identify the parameter set with the fewest iterations to convergence
- Verify final energies are consistent across parameter sets
Interpretation: Lower iteration counts indicate better performance, but consistent final energies must be confirmed to ensure physical validity.
Protocol 2: Advanced Parameter Optimization for Challenging Systems
Purpose: To address systems that fail to converge with standard parameter screening.
Materials and Setup:
- As in Protocol 1, plus:
- Access to SCF acceleration methods (Broyden, LIST, MESA)
- Electron smearing capabilities
- Level shifting options
Procedure:
- If oscillations occur, reduce mixing weight by 50% and implement Pulay mixing with increased history length (4-8) [33]
- For persistent oscillations, switch to Broyden method with similar parameters [33]
- For metallic systems or those with small HOMO-LUMO gaps:
- For magnetic systems:
- As a last resort for extremely difficult cases:
Interpretation: Successful convergence should show monotonic decrease in energy and density/Hamiltonian changes. Consistent final energies across different methods validate the result.
Protocol 3: DOS-Specific Convergence Verification
Purpose: To ensure SCF convergence is adequate for accurate density of states calculations.
Background: DOS calculations often require higher k-point sampling and more stringent convergence criteria than total energy calculations [35]. The relationship between k-point sampling and DOS quality involves multiple factors: Brillouin zone integration scheme, k-point sampling fineness, energy grid fineness, DOS smoothing, and band dispersion [35].
Materials and Setup:
- Converged geometry from previous calculations
- Fine k-point grid appropriate for DOS calculations [35]
- High-energy cutoff if using plane-wave basis
Procedure:
- First, converge the system using Protocols 1 or 2 with standard k-point grid
- Increase k-point density by at least 2× in each direction for final DOS calculation [35]
- Use the optimal mixing parameters identified in step 1
- Verify convergence with tighter criteria (e.g.,
SCF.DM.Tolerance = 10⁻⁵) [33] - Calculate DOS using tetrahedron method or appropriate smearing [35]
- Check DOS reproducibility with slightly different mixing parameters
Interpretation: The DOS should be stable with respect to small changes in mixing parameters and SCF convergence criteria.
Table 2: Essential Computational Tools for SCF Convergence Research
| Tool/Resource | Function/Purpose | Implementation Examples |
|---|---|---|
| Pulay/DIIS Mixer | Accelerates SCF convergence using history of previous steps [33] | SIESTA: SCF.Mixer.Method Pulay [33]; ADF: Default DIIS [2] |
| Broyden Mixer | Alternative acceleration method; sometimes superior for metallic systems [33] | SIESTA: SCF.Mixer.Method Broyden [33]; ABACUS: Default method [36] |
| Electron Smearing | Enables fractional occupancies for metallic systems [32] [36] | ABACUS: smearing_method and smearing_sigma [36]; ADF: Occupations settings [32] |
| MESA Algorithm | Combines multiple acceleration methods for difficult cases [2] [32] | ADF: AccelerationMethod MESA [2] |
| Level Shifting | Artificially raises virtual orbital energies to aid convergence [32] | ADF: Lshift parameter (enables OldSCF) [32] |
| Adaptive History Length | Controls how many previous steps are used in Pulay/Broyden [33] | SIESTA: SCF.Mixer.History [33]; ADF: DIIS N [2] |
Optimal configuration of SCF mixing parameters is system-dependent and crucial for efficient and accurate electronic structure calculations. Small molecules typically perform well with default parameters, while metallic, magnetic, and open-shell systems require more careful parameterization. For DOS calculations, which demand high accuracy in the electronic structure, verifying convergence with respect to mixing parameters is particularly important.
The protocols provided herein offer systematic approaches for determining optimal parameters across diverse molecular systems. By following these guidelines and understanding the underlying principles of SCF convergence, researchers can significantly improve the efficiency and reliability of their computational workflows, especially in the context of DOS convergence research for energy grid parameterization.
This application note provides detailed protocols for implementing two advanced computational techniques essential for researchers conducting electronic structure calculations, particularly in the context of density of states (DOS) convergence research. Achieving converged results in computational materials science and drug development requires sophisticated approaches to handle temperature effects and optimization processes. We focus specifically on finite electronic temperature methodologies, which account for thermal effects on electronic properties, and adaptive convergence criteria, which dynamically control iterative solvers to improve computational efficiency. These techniques are particularly valuable for setting energy grid parameters in DOS calculations where accuracy and computational cost must be carefully balanced.
The content is structured to provide immediately applicable knowledge, featuring comparative tables of methodological approaches, detailed experimental protocols, visualization of computational workflows, and essential research toolkits. This framework supports researchers in materials science and computational drug development who require robust methods for predicting material properties and behavior at finite temperatures.
Finite Electronic Temperature Methodology
Theoretical Foundation and Computational Approaches
Incorporating finite electronic temperature is crucial for simulating realistic material behavior, as it accounts for how thermal excitations influence electronic structure, magnetic properties, and transport phenomena. For Density of States (DOS) convergence research, this is particularly important as temperature effects can significantly alter electronic distributions near the Fermi level.
Table 1: Comparison of Finite-Temperature Simulation Approaches for DOS Calculations
| Method | Key Principle | Temperature Treatment | Best Suited Materials | Computational Cost |
|---|---|---|---|---|
| Classical Heisenberg Model with Boltzmann distribution | Treats spin moments as classical vectors [37] | Boltzmann distribution for thermal fluctuations | General ferromagnets near TC | Medium |
| Quantum-Corrected Approach with Bose-Einstein statistics | Incorporates magnon quantization effects [37] | Bose-Einstein distribution for magnon excitations | bcc Fe, other ferromagnets at low T | High |
| First-Principles with Thermal Lattice Vibrations | DFT-derived Jij with thermal lattice effects [37] | CPA averaging of atomic displacements | Systems with strong electron-phonon coupling | Very High |
| Monte Carlo with Quantum Fluctuation-Dissipation | Modified Monte Carlo sampling [37] | Quantum fluctuation-dissipation ratio ηqt(T) | Low-temperature magnetic systems | High |
For DOS calculations, the finite-temperature electronic structure forms the foundation for understanding various material properties. As emphasized in electronic structure analysis, "The density of states of electrons is a simple, yet highly-informative, summary of the electronic structure of a material" [38]. When temperature effects are properly incorporated, researchers can more accurately predict effective mass, Van Hove singularities, and the effective dimensionality of electrons in materials.
Protocol: Implementing Finite Electronic Temperature in DOS Calculations
Objective: To incorporate finite electronic temperature effects in density of states calculations for body-centered cubic (bcc) iron, enabling more accurate prediction of magnetic and transport properties.
Materials and Computational Resources:
- First-principles calculation software (VASP, SPR-KKR)
- Monte Carlo simulation framework for magnetic systems
- High-performance computing resources
- Post-processing tools for DOS analysis
Procedure:
Initial Structure Relaxation
- Perform crystal structure relaxation using DFT code (e.g., VASP) with GGA-PBE functional [37]
- Utilize a k-point mesh of 24×24×24 and plane-wave basis set cutoff energy of 500 eV
- Confirm convergence of structural parameters to tolerances of 0.01 eV for energy and 0.01 eV/Å for forces
Phonon Calculations for Thermal Lattice Effects
- Compute force constants using density functional perturbation theory
- Employ supercells of varying sizes (2×2×2, 3×3×3, and 4×4×4) for comprehensive sampling
- Derive phonon dispersion curves and phonon density of states using Phonopy code [37]
- Calculate specific heat of the lattice using harmonic approximations
Temperature-Dependent Exchange Coupling Constants
- Evaluate magnetic exchange coupling constants (Jij) using Liechtenstein formula within SPR-KKR code
- Incorporate thermal lattice vibration effects through coherent potential approximation (CPA)
- Generate multiple atomic displacement configurations characterized by probabilities xv for v=1,…,Nv [37]
- Compute Jij over temperature range from 0 K to 1600 K with 25 K intervals
Monte Carlo Simulations with Quantum Corrections
- Implement classical Heisenberg model with temperature-dependent exchange interactions
- Apply quantum fluctuation-dissipation ratio ηqt(T) to account for Bose-Einstein statistics of magnons: ηqt(T) = ∫[ℏω/2 + (ℏω)/(e^(ℏω/kBT) - 1)]gm(ω,T)dω [37]
- Execute Monte Carlo sampling with modified probability distribution: P({Si}) = C × exp(-E/η(T))
- Calculate magnetization and magnetic energy as functions of lattice temperature
Finite-Temperature DOS Calculation
- Compute electronic structures while accounting for both thermal lattice vibration and thermal spin fluctuation effects
- Utilize the Kubo-Greenwood formula for temperature-dependent electrical resistivity as validation
- Confirm that spontaneous magnetization follows Bloch's T^(3/2) power law in low-temperature regime
Validation:
- Compare calculated Curie temperature with experimental value (1043 K for bcc Fe)
- Verify that spontaneous magnetization matches experimental curves across temperature range
- Confirm electrical resistivity trends align with experimental measurements
Finite Temperature DOS Calculation Workflow
Adaptive Convergence Criteria
Theoretical Framework and Method Selection
Adaptive convergence criteria dynamically control iterative processes in computational simulations, balancing solution accuracy with computational expense. For DOS convergence research, appropriate convergence criteria are essential for obtaining reliable results without excessive computational overhead.
Table 2: Adaptive Convergence Criteria for Iterative Methods
| Criterion Type | Mathematical Formulation | Applications | Advantages | Limitations |
|---|---|---|---|---|
| Absolute Difference | ⎸V(Xt) - V(Xt+1)⎹ ≤ μ(1-β)/2β [39] | SDP for reservoir systems | Simple implementation | May converge slowly for flat value functions |
| Squared Difference | ∑(V(Xt) - V(Xt+1))² ≤ μ(1-β)/2β [39] | Multi-reservoir optimization problems | Faster convergence for smooth functions | More sensitive to outliers |
| Adaptive Grid Refinement | Metric-based cell subdivision [40] | CFD, unstructured hexahedral grids | Automatic focus on high-error regions | Complex implementation |
| Dual Certificate Violation | Branch-and-bound detection [41] | Total variation minimization | Avoids heuristic approaches | Requires dual problem formulation |
The fundamental challenge in convergence criterion selection lies in the balance between computational efficiency and solution accuracy. As noted in reservoir optimization studies, "an incremental solution strategy based on an iterative method will be effective if, and only if, the selection of the convergence criterion is adequate for the completion of the iteration process" [39]. Adaptive grid refinement methods have demonstrated particular value for complex systems where "the grid resolution needed to resolve the flow phenomena, as well as the precise position of these features, is uncertain" [40].
Protocol: Implementing Adaptive Convergence for DOS Calculations
Objective: To implement and validate adaptive convergence criteria for density of states calculations, reducing computational time while maintaining required accuracy.
Materials and Computational Resources:
- Electronic structure calculation software
- Grid refinement capabilities (isotropic and anisotropic)
- Programming environment for algorithm customization
- Convergence monitoring tools
Procedure:
Baseline Convergence Parameter Establishment
- Set initial tolerance value μ = 10⁻³ as reference point [39]
- Select discount factor β between 0 and 1 (typically 0.95-0.99)
- Determine maximum iteration count based on system complexity and computational constraints
Convergence Criterion Implementation
- Option A: Absolute Difference Criterion
- Calculate D = ∑⎸V(Xt) - V(Xt+1)⎹ across all states [39]
- Compare against threshold μ(1-β)/2β
- Option B: Squared Difference Criterion
- Calculate D = ∑(V(Xt) - V(Xt+1))² across all states [39]
- Compare against threshold μ(1-β)/2β
- For grid-based DOS calculations, implement metric-based adaptive refinement:
- Option A: Absolute Difference Criterion
Adaptive Refinement Process
- For initial iterations, use coarser convergence criteria to accelerate early progress
- Implement conditional refinement based on dual constraint violations for total variation minimization [41]
- Gradually tighten criteria as solution approaches convergence
- For DOS calculations, focus refinement on regions near Fermi level where higher resolution is critical
Convergence Monitoring and Dynamic Adjustment
- Track convergence rate throughout iterative process
- If oscillation detected, increase damping factor or adjust refinement aggressiveness
- For stochastic methods like Wang-Landau, modify the density of states estimate using distribution of random walkers without auxiliary modification factors [42]
Validation and Accuracy Assessment
- Compare results obtained with adaptive criteria against reference calculations with stringent fixed criteria
- Verify that key DOS features (band edges, Van Hove singularities) are preserved
- Quantify computational time savings and any accuracy trade-offs
Troubleshooting:
- For oscillatory behavior: Increase discount factor β or implement moving average smoothing
- For premature convergence: Tighten tolerance μ or implement additional validation checks
- For excessive computation time: Loosen criteria in well-behaved regions or implement multi-level approach
Adaptive Convergence Decision Process
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational Tools for Finite Temperature and Convergence Research
| Tool/Category | Specific Examples | Function/Purpose | Application Context |
|---|---|---|---|
| First-Principles Codes | VASP, SPR-KKR [37] | Electronic structure calculation | DFT-based property prediction |
| Monte Carlo Frameworks | Custom Heisenberg model implementations [37] | Statistical sampling of configurations | Finite temperature magnetism |
| Phonon Calculators | Phonopy [37] | Lattice vibration analysis | Thermal lattice effects |
| Adaptive Mesh Tools | ISIS-CFD, custom refinement algorithms [40] [41] | Grid optimization | Resolution enhancement in critical regions |
| Convergence Monitors | Custom convergence criteria scripts [39] | Iteration control | Computational efficiency |
| Temperature Control | PT100 thermal resistors, magneto-electric angle sensors [43] | Experimental validation | Benchmarked comparison |
| Neural Network | GA-BP neural network [44] | Temperature prediction | Device thermal safety |
This application note has detailed protocols for implementing finite electronic temperature methodologies and adaptive convergence criteria in DOS convergence research. The finite temperature approach, particularly through quantum-corrected Monte Carlo methods, enables more accurate prediction of material properties across temperature ranges relevant to operational conditions. The adaptive convergence criteria provide researchers with structured approaches to balance computational efficiency with solution accuracy, essential for complex DOS calculations where a priori knowledge of required resolution is limited.
Together, these advanced techniques empower researchers to conduct more realistic simulations of materials and molecular systems, with particular value for drug development professionals investigating temperature-dependent biomolecular interactions and materials scientists designing novel functional materials. The provided protocols, visualization workflows, and research toolkit facilitate immediate implementation and adaptation to diverse research scenarios.
Selecting an appropriate atomic orbital basis set represents a fundamental and critical decision in electronic structure calculations, particularly for research focused on setting energy grid parameters for Density of States (DOS) convergence. The principal challenge lies in navigating the inherent trade-off between computational accuracy and efficiency—a dilemma known as the "conundrum of diffuse basis sets" where researchers must balance the blessing of accuracy against the curse of sparsity [45]. This balance is especially crucial in DOS convergence research, where the precise characterization of electronic energy levels demands basis sets capable of accurately representing both localized and diffuse electron densities without making computations prohibitively expensive.
The essential compromise revolves around basis set completeness: smaller basis sets offer computational tractability but introduce significant basis set incompleteness error (BSIE) and basis set superposition error (BSSE), while larger basis sets provide superior accuracy but dramatically increase computational costs and can adversely affect the sparsity of the one-particle density matrix [45] [46]. For DOS convergence studies, where the accurate representation of both occupied and virtual orbitals is essential, this selection process becomes paramount, as the choice directly influences the convergence behavior, cost-effectiveness, and ultimate reliability of the calculated electronic properties.
Theoretical Foundation: The Accuracy-Sparsity Conundrum
The Mathematical Basis of the Trade-off
The fundamental challenge in basis set selection arises from competing mathematical properties of the one-particle density matrix (1-PDM). For insulating systems with significant HOMO-LUMO gaps, matrix elements of the 1-PDM are expected to decay exponentially with increasing real-space distance from the diagonal, suggesting inherent sparsity that could enable linear-scaling electronic structure methods [45]. However, this theoretical sparsity is severely compromised when using diffuse basis sets, creating the central conundrum: while diffuse functions are absolutely essential for accurate description of non-covalent interactions, they have a "detrimental impact on the sparsity of the 1-PDM" that extends beyond what the spatial extent of the basis functions alone would predict [45].
This mathematical behavior manifests as a "curse of sparsity" where the inverse overlap matrix (S⁻¹) exhibits significantly lower locality than its co-variant dual, creating numerical challenges that persist even after projecting the 1-PDM onto a real-space grid [45]. Counterintuitively, this sparsity problem worsens with larger basis sets, seemingly contradicting the expectation of a well-defined basis set limit. Research has shown this effect is proportional to both the diffuseness and local incompleteness of the basis set, meaning "small and diffuse basis sets are affected the most" [45].
Implications for DOS Convergence Research
For Density of States convergence studies, these mathematical properties have direct practical implications. The compromised sparsity of the 1-PDM when using diffuse basis sets translates to later onset of the low-scaling regime, larger cutoff errors from sparse treatment, and sometimes erratic convergence behavior [45]. This creates particular challenges for DOS calculations on larger systems or those requiring extensive sampling of the Brillouin zone, where computational efficiency becomes as important as accuracy.
The relationship between basis set quality and DOS convergence is nonlinear—initial improvements from increasing basis set size yield significant gains in DOS accuracy, but this progression eventually reaches a point of diminishing returns where further basis set expansion provides minimal improvement at excessive computational cost. Identifying the optimal point on this curve is essential for efficient DOS convergence research.
Quantitative Analysis of Basis Set Performance
Table 1: Accuracy and Performance Trade-offs Across Basis Set Families for Non-Covalent Interactions
| Basis Set | NCI RMSD (M+B) (kJ/mol) | Relative Computation Time | Recommended Application |
|---|---|---|---|
| def2-SVP | 31.51 | 1.0× | Preliminary screening |
| def2-TZVP | 8.20 | 3.2× | Geometry optimization |
| def2-TZVPPD | 2.45 | 9.5× | Final NCI calculations |
| cc-pVDZ | 30.31 | 1.2× | Large system screening |
| cc-pVTZ | 12.73 | 3.8× | General purpose |
| aug-cc-pVDZ | 4.83 | 6.5× | Moderate-accuracy NCI |
| aug-cc-pVTZ | 2.50 | 17.9× | High-accuracy NCI |
| aug-cc-pVQZ | 2.40 | 48.3× | Benchmark quality |
Data adapted from ASCDB benchmark studies using ωB97X-V functional [45]
Table 2: Performance of Double-ζ Basis Sets Across Multiple Functionals (WTMAD2 Values from GMTKN55)
| Basis Set | B97-D3BJ | r2SCAN-D4 | B3LYP-D4 | M06-2X | ωB97X-D4 |
|---|---|---|---|---|---|
| def2-QZVP | 8.42 | 7.45 | 6.42 | 5.68 | 3.73 |
| vDZP | 9.56 | 8.34 | 7.87 | 7.13 | 5.57 |
| def2-SVP | 14.92 | 12.60 | 11.45 | 10.84 | 8.91 |
| 6-31G(d) | 16.53 | 14.18 | 13.02 | 12.47 | 10.26 |
| pcseg-1 | 13.71 | 11.25 | 10.18 | 9.67 | 7.89 |
Lower WTMAD2 values indicate better accuracy. Data from Wagen and Vandezande (2024) [46]
The quantitative data reveals several critical patterns for DOS convergence research. First, the addition of diffuse functions provides dramatic improvements for non-covalent interactions, with def2-TZVPPD reducing errors by approximately 70% compared to def2-TZVP while increasing computational time by roughly 3× [45]. Second, the vDZP basis set emerges as a particularly efficient double-ζ option, delivering accuracy much closer to triple-ζ basis sets while maintaining the computational cost characteristic of double-ζ basis sets [46]. Third, the performance gap between conventional double-ζ basis sets (like def2-SVP) and larger basis sets is especially pronounced for non-covalent interactions, highlighting the importance of basis set selection for properties dependent on weak interactions.
Basis Set Selection Protocol for DOS Convergence Studies
Protocol Implementation Guidelines
The basis set selection workflow begins with clearly defining research objectives, as this determines the required accuracy level and appropriate starting point in the protocol. For rapid screening of large molecular databases or preliminary conformational analysis, compact double-ζ basis sets like vDZP or def2-SVP provide the best efficiency [46]. For moderate accuracy requirements including most geometry optimizations and frequency calculations, standard triple-ζ basis sets without diffuse functions (def2-TZVP, cc-pVTZ) typically offer the optimal balance [45] [47]. For high-accuracy applications such as final single-point energies, reaction barriers, or properties dependent on non-covalent interactions, augmented triple-ζ basis sets (def2-TZVPPD, aug-cc-pVTZ) are essential [45].
System size considerations directly impact practical feasibility. For systems exceeding 100 atoms, compact basis sets like vDZP or pcseg-n families are strongly recommended due to their favorable scaling properties and reduced incidence of linear dependence [46]. Medium-sized systems (20-100 atoms) can typically accommodate standard triple-ζ basis sets, while small systems (<20 atoms) can exploit the full accuracy of augmented correlation-consistent basis sets, potentially up to quintuple-ζ for ultimate convergence testing [45] [47].
Element-specific considerations must be addressed—heavy elements require specialized basis sets with appropriate effective core potentials or core-valence correlation consistency. For non-covalent interactions, diffuse functions are "absolutely essential for an accurate description" [45], while general organic molecules perform well with standard polarized triple-ζ basis sets.
Specialized Protocol for DOS Convergence Research
For Density of States convergence studies, implement this specialized protocol:
- Initial Assessment: Begin with a moderate-sized triple-ζ basis set (def2-TZVP, cc-pVTZ) for initial DOS calculations to establish baseline convergence behavior.
- Diffuse Function Testing: Compare results with augmented basis sets (def2-TZVPPD, aug-cc-pVTZ) to assess the sensitivity of virtual orbitals and conduction band features to diffuse functions.
- Basis Set Extrapolation: For highest accuracy, employ a systematic series of basis sets (e.g., cc-pVXZ where X = D, T, Q) and extrapolate to the complete basis set limit using established extrapolation formulae.
- Compact Basis Set Validation: Test specialized compact basis sets (vDZP, pcseg-n) against larger standards to determine if they provide sufficient accuracy for production calculations.
- Sparsity Assessment: For large systems, evaluate the sparsity of the one-particle density matrix when using diffuse functions, as this directly impacts computational feasibility [45].
Research Reagent Solutions: Essential Computational Tools
Table 3: Essential Basis Sets and Their Applications in DOS Research
| Basis Set | Type | Primary Applications | Key Features | Limitations |
|---|---|---|---|---|
| vDZP | Compact DZ | Large system screening, High-throughput studies | Minimal BSSE, Optimized contractions, Effective core potentials | Limited flexibility for anisotropic densities |
| def2-SVP | Standard DZ | Preliminary geometry optimization, Molecular dynamics | Balanced cost/accuracy, Wide element coverage | Significant BSIE for properties |
| def2-TZVP | Standard TZ | Production geometry optimization, Frequency calculation | Excellent balance for most applications, Good element coverage | Lacks diffuse functions for NCIs |
| def2-TZVPPD | Augmented TZ | Non-covalent interactions, Reaction barriers, Accurate DOS | Diffuse functions added, Excellent for weak interactions | Increased computational cost |
| cc-pVDZ | Correlation-consistent DZ | Initial CBS extrapolation, Educational applications | Systematic improvement path, Excellent for CBS extrapolation | Requires large X for accuracy |
| cc-pVTZ | Correlation-consistent TZ | High-accuracy single-point, Benchmark studies | Systematic construction, Excellent for electron correlation | Computational cost for large systems |
| aug-cc-pVTZ | Augmented correlation-consistent TZ | Benchmark NCIs, Spectroscopic properties, Final DOS | Comprehensive diffuse functions, Superior for excited states | High computational cost, Linear dependence issues |
| pcseg-n | Polarization-consistent | DFT specialization, Property calculation | Optimized for DFT, Good performance/cost balance | Less common for wavefunction methods |
Implementation Notes for Research Reagents
The vDZP basis set deserves special consideration as it represents a recent advancement in compact basis set design. Its deeply contracted valence basis functions are "optimized on molecular systems to minimize BSSE almost down to the triple-ζ level" [46], making it particularly valuable for DOS convergence studies on large systems where computational efficiency is paramount. Validation studies demonstrate that vDZP-based methods "have speed and accuracy similar to existing composite methods" while "substantially outperforming conventional double-ζ basis sets" [46].
For correlation-consistent basis sets, the systematic construction allows for reliable complete basis set (CBS) extrapolation, which is especially valuable for DOS convergence research where estimating the complete basis set limit is often a primary objective. The augmentation with diffuse functions in the aug-cc-pVXZ series is particularly important for accurately characterizing unoccupied orbitals and conduction band states in DOS calculations.
The def2 family offers practical advantages for general-purpose research, with consistent design across the periodic table and availability in most quantum chemistry software packages. The def2-TZVPPD level provides an excellent compromise for DOS studies requiring accurate treatment of weak interactions without the full cost of aug-cc-pVQZ or larger basis sets.
Experimental Protocols for Basis Set Validation
Protocol 1: DOS Convergence Assessment
Objective: Quantitatively evaluate basis set convergence for Density of States calculations.
Procedure:
- Select a series of basis sets of increasing quality (e.g., cc-pVDZ → cc-pVTZ → cc-pVQZ or def2-SVP → def2-TZVP → def2-QZVP)
- Perform single-point energy calculations with consistent functional and integration grid settings
- Calculate the DOS for each basis set using consistent broadening parameters
- Quantify convergence by monitoring:
- Position and spacing of key spectral features
- HOMO-LUMO gap convergence
- Integral of DOS up to specific energy thresholds
- Root-mean-square deviation between successive basis set levels
Validation Metrics:
- Total energy change < 1 mHa between successive basis set levels
- HOMO-LUMO gap variation < 0.01 eV
- Spectral feature positions stable within 0.05 eV
Protocol 2: Computational Cost Assessment
Objective: Measure computational scaling with basis set size for system-specific resource planning.
Procedure:
- Select representative molecular structures spanning expected system sizes
- Perform timing studies with consistent computational resources
- Measure computation time for key steps: SCF convergence, integral evaluation, and property calculation
- Fit scaling coefficients to establish empirical computational cost model
Analysis:
- Compare observed scaling with theoretical expectations
- Identify threshold system sizes where computational demands become prohibitive
- Establish practical basis set limits for different research phases
Emerging Solutions and Future Directions
Addressing the Sparsity Challenge
Recent investigations into the sparsity problem presented by diffuse basis sets have identified promising approaches. The complementary auxiliary basis set (CABS) singles correction in combination with compact, low quantum-number basis sets shows potential for mitigating the sparsity problem while maintaining accuracy for non-covalent interactions [45]. This approach addresses the fundamental issue that the "inverse overlap matrix being significantly less sparse than its co-variant dual" [45], which underlies the numerical challenges with diffuse functions.
For DOS convergence research, specialized basis set strategies that separate the treatment of occupied and virtual orbital spaces may offer improved efficiency. Such approaches could provide the diffuse functions necessary for accurate characterization of unoccupied states in DOS calculations while maintaining better sparsity properties for the occupied orbital space.
Method-Specific Optimization
The development of method-specific basis sets continues to provide efficiency gains. The success of the vDZP basis set across multiple density functionals demonstrates that "specially optimized combinations of functionals, basis sets, and empirical corrections" can deliver "robustness and computational efficiency" without method-specific reparameterization [46]. This suggests similar opportunities exist for developing DOS-specialized basis sets optimized for the specific requirements of density of states calculations, potentially focusing on accurate representation of both valence and low-lying virtual orbitals with minimal overall basis set size.
Future directions will likely include increased use of machine learning approaches to develop system-specific adaptive basis sets that dynamically adjust to molecular environment, potentially offering both improved accuracy and computational efficiency for DOS convergence studies across diverse chemical systems.
Step-by-Step Protocol for Initial Parameter Setup in Drug-like Molecules
Setting initial parameters for drug-like molecules is a critical first step in computational drug discovery, directly influencing the accuracy and reliability of subsequent simulations and predictions. This protocol focuses on establishing robust initial energy grid parameters and computational specifications to ensure density of states (DOS) convergence—a fundamental requirement for obtaining physically meaningful results in electronic structure calculations. The parameterization framework outlined here is grounded in first-principles quantum mechanics and leverages benchmarked datasets to achieve optimal balance between computational efficiency and predictive accuracy. Within the broader context of DOS convergence research, proper initialization ensures that sampled molecular configurations adequately represent the chemical space of drug-like compounds, enabling more reliable virtual screening and binding affinity predictions in structure-based drug design campaigns.
Computational Specifications & Theoretical Foundation
Reference Quantum Chemical Method
The recommended reference method for target calculations is ωB97M-D3(BJ)/def2-TZVPPD, which has been extensively validated for drug-like molecules [48]. This hybrid meta-GGA density functional with dispersion correction provides an excellent compromise between computational cost and accuracy, particularly for non-covalent interactions prevalent in biological systems. The def2-TZVPPD basis set offers enhanced flexibility for describing conformational landscapes and electronic properties while maintaining reasonable computational requirements for molecules of pharmaceutical relevance.
Key Dataset for Parameterization
The QDπ dataset serves as the primary reference for parameter validation, containing 1.6 million structures encompassing 13 biologically relevant elements [48]. This dataset incorporates diverse molecular motifs including conformational isomers, transition states, intermolecular complexes, tautomers, and protonation states specifically relevant to drug discovery. The chemical space coverage ensures that parameter initialization accounts for the diverse electronic environments encountered in pharmaceutical compounds.
Table 1: Benchmark Dataset for Parameter Validation
| Dataset | Structures | Elements | Theory Level | Special Features |
|---|---|---|---|---|
| QDπ | 1.6 million | 13 | ωB97M-D3(BJ)/def2-TZVPPD | Drug-like molecules, biopolymer fragments |
| SPICE | ~1.1 million | 6 | ωB97M-D3(BJ)/def2-TZVPPD | Biologically relevant molecules |
| ANI-2x | ~8.6 million | 8 | ωB97X/6-31G* | Broad chemical space |
Step-by-Step Parameter Setup Protocol
System Preparation and Initialization
Step 1: Molecular Structure Preparation
- Obtain 3D molecular structures from reliable sources (Protein Data Bank, ZINC, or ChEMBL)
- Ensure proper protonation states at physiological pH (7.4) using tools like OpenBabel or RDKit
- Generate initial conformers using distance geometry methods with constraints for chiral centers
- For flexible molecules with >5 rotatable bonds, employ systematic conformer search with energy window of 10-15 kcal/mol
Step 2: Basis Set Selection
- Select def2-TZVPPD as the primary basis set for all elements (H, C, N, O, F, P, S, Cl, and biologically relevant metals)
- Implement effective core potentials for transition metals when present
- Ensure consistent basis set treatment across all calculations to maintain energy consistency
Step 3: Integration Grid Specification
- Set radial grid points to 150 for elements H-C-N-O and 250 for heavier elements
- Configure angular grid to Lebedev-Laikov scheme with 770 points for accurate numerical integration
- Specify grid spacing of 0.15-0.20 Bohr for real-space representation of electron density
- For DOS calculations, set energy range from -50 eV to +50 eV relative to Fermi level with resolution of 0.01 eV
Convergence Parameter Configuration
Step 4: SCF Convergence Criteria
- Set energy convergence threshold to 10⁻⁸ Hartree for high-precision requirements
- Configure density matrix convergence to 10⁻⁷ for stable DOS calculations
- Implement DIIS (Direct Inversion in Iterative Subspace) accelerator with initial guess from atomic superposition
- For difficult-to-converge systems, employ damping factor of 0.2 and increase to 0.5 if necessary
Step 5: DOS-Specific Parameters
- Set k-point sampling to Γ-point only for molecular systems
- Configure Gaussian smearing width of 0.05-0.10 eV for state occupancy
- Specify energy grid for DOS output with minimum 5000 points across selected energy range
- Enable projected DOS (PDOS) for orbital analysis with atom-centered projections
Step 6: Geometry Optimization Pre-Processing
- Set initial force tolerance to 0.05 eV/Å for preliminary relaxation
- Configure energy change tolerance to 10⁻⁵ Hartree between optimization steps
- Select L-BFGS (Limited-memory Broyden-Fletcher-Goldfarb-Shanno) optimizer for systems with >50 atoms
- Implement internal coordinate system for flexible molecules with rotatable bonds
Table 2: Critical Convergence Parameters for DOS Calculations
| Parameter | Initial Value | Converged Value | Threshold for Drug-like Molecules |
|---|---|---|---|
| SCF Energy Tolerance | 10⁻⁶ Hartree | 10⁻⁸ Hartree | 10⁻⁷ Hartree |
| Force Tolerance | 0.05 eV/Å | 0.01 eV/Å | 0.015 eV/Å |
| DOS Energy Grid | 1000 points | 5000 points | 2000 points |
| k-point Sampling | 1×1×1 | 3×3×3 (periodic) | Γ-point (molecular) |
| Smearing Width | 0.20 eV | 0.05 eV | 0.10 eV |
Workflow Diagram for Parameter Setup
Validation and Quality Control Protocol
Active Learning Validation
Implement a query-by-committee active learning strategy to validate parameter settings [48]. This approach utilizes multiple independent models to identify regions of chemical space where predictions diverge, indicating insufficient parameter convergence:
- Train 4 independent machine learning potentials with different random seeds
- Calculate energy and force standard deviations across committee members
- Flag structures with standard deviations >0.015 eV/atom for energy or >0.20 eV/Å for forces
- Iteratively refine parameters until all committee members agree within thresholds
- Use DP-GEN software implementation for automated active learning cycles
Reference Data Comparison
Validate calculated properties against benchmark data from the QDπ dataset:
- Compare conformational energies for flexible drug-like molecules
- Validate intermolecular interaction energies for protein-ligand complexes
- Assess tautomerization and protonation state energies
- Verify electronic properties including HOMO-LUMO gaps and orbital distributions
- Check density of states convergence against high-level reference calculations
Research Reagent Solutions
Table 3: Essential Computational Tools for Parameter Setup
| Tool/Software | Function | Application Context |
|---|---|---|
| PSI4 v1.7+ | Quantum Chemistry Package | Reference ωB97M-D3(BJ) calculations [48] |
| DP-GEN | Active Learning Framework | Parameter validation and refinement [48] |
| AutoDock | Molecular Docking | Binding pose prediction and scoring [49] |
| DOCK3.7 | Structure-Based Screening | Large-scale virtual screening [50] |
| FREED++ | Reinforcement Learning | Generative molecular design [51] |
| Glide | Precise Docking | Induced-fit and flexible docking [49] |
| GOLD | Genetic Algorithm Docking | Protein-ligand pose prediction [49] |
Advanced Implementation Notes
Machine Learning Potential Integration
For large-scale screening applications, consider integrating semiempirical quantum mechanical (SQM)/Δ MLP models [48]. This approach combines the computational efficiency of semiempirical methods with the accuracy correction of machine learning potentials:
- Use GFN2-xTB or PM7 as baseline semiempirical methods
- Train Δ-MLP to reproduce difference between SQM and ωB97M-D3(BJ)/def2-TZVPPD
- Implement short-range non-electrostatic corrections for condensed phase simulations
- Validate transferability across diverse drug-like chemical space
Performance Optimization
- Utilize resolution-of-identity (RI) approximation for two-electron integrals to accelerate calculations
- Implement parallelization over multiple k-points for periodic systems
- Employ GPU acceleration for density functional computations
- Use hierarchical data compression for large-scale DOS storage and analysis
This protocol provides a comprehensive framework for initial parameter setup specifically tailored to drug-like molecules, with emphasis on DOS convergence requirements. Proper implementation of these steps establishes a robust foundation for accurate and efficient computational drug discovery workflows.
Advanced Troubleshooting: Diagnosing and Resolving Persistent SCF Convergence Failures
In the context of research on setting energy grid parameters for Denial-of-Service (DOS) convergence, the ability to systematically diagnose oscillations is paramount. Oscillations in grid components, such as control loops, can propagate, leading to system-wide instability, performance degradation, and even failure to converge to a stable operating state [52] [53]. The integration of volatile elements like Electric Vehicle (EV) charging infrastructure introduces new layers of complexity, making robust diagnostic protocols essential for maintaining grid reliability [54]. This document provides detailed application notes and experimental protocols for identifying the root causes of oscillation and divergence in such systems, with a particular focus on applications within modern energy grids.
Performance data from recent studies demonstrates the impact of advanced optimization and the consequences of oscillation in control systems.
Table 1: Performance Metrics of an EV-Integrated Grid Optimization Model This table summarizes key results from a multi-objective optimization framework for a 33-bus distribution grid, showcasing the potential performance gains from effective management. All data is benchmarked against a scenario without EV integration [54].
| Performance Metric | Improvement with EV Integration | Benchmark Comparison |
|---|---|---|
| Operational Cost | Reduced by 19.3% | 4.4% lower cost than Komodo Mlipir Algorithm (KMA) |
| Energy Losses | Decreased by 59.7% | 24.5% lower loss than Particle Swarm Optimization (PSO) |
| Load Shedding | Minimized by 75.4% | - |
| Voltage Deviations | Improved by 43.5% | - |
| PV Curtailment | Eliminated | - |
Table 2: Common Causes of Oscillations in Industrial Control Loops This table categorizes the primary root causes of oscillations in linear closed-loop systems, as identified in process control literature. A single loop may be affected by one or multiple of these causes simultaneously [52].
| Root Cause Category | Specific Cause | Typical Origin |
|---|---|---|
| Component Nonlinearity | Control valve stiction or deadband | Leads to limit cycles in a control loop [52] |
| Controller Tuning | Poorly tuned or marginally stable controller | Destabilizes the system, especially after process changes [52] |
| External Disturbance | Propagating oscillatory disturbance | Originates from interactions with other loops or external factors [52] |
Experimental Protocols for Oscillation Diagnosis
Protocol: Integrated Oscillation Root Cause Analysis (RCA) for Linear SISO Systems
This protocol provides a methodology to diagnose single or multiple root causes for oscillations in linear Single-Input-Single-Output (SISO) systems, integrating several techniques for a comprehensive analysis [52].
- 1.0 Objective: To identify the presence and type of root causes—including valve stiction, aggressively tuned controllers, and external oscillatory disturbances—for oscillations in linear closed-loop systems.
- 2.0 Prerequisites:
- Time-series data of the Process Variable (PV) and Controller Output (OP/CO).
- Access to oscillation detection and characterization tools.
- A validated stiction detection model (e.g., Hammerstein-based).
- 3.0 Procedure:
- Data Collection & Preprocessing: Collect historical data for the PV and OP signals from the control loop of interest. Preprocess the data to remove noise and outliers if necessary.
- Oscillation Detection & Characterization: Apply a multiple oscillation detection algorithm (e.g., as described in Babji and Rengaswamy, 2011) to the PV data. Determine the number of distinct oscillatory components present.
- Single vs. Multiple Cause Determination: If the characterization algorithm detects only one oscillatory component, proceed with a single root cause analysis. If multiple oscillatory components are detected, the system is likely experiencing multiple simultaneous root causes.
- Stiction Detection Check: a. Apply a Hammerstein model-based stiction detection method to the PV and OP data. b. If stiction is confirmed and only a single oscillation is present, stiction is the root cause. c. If stiction is confirmed but multiple oscillations are present, stiction is one of multiple root causes.
- Discrimination Between Tuning and Disturbance: For loops where stiction is not detected, use an amplitude-based discrimination algorithm. a. Analyze the oscillation's characteristics using methods like Hilbert-Huang (HH) spectrum analysis. b. If the oscillation's amplitude is consistent with a specific pattern, it may indicate a marginally stable loop due to aggressive tuning. c. Otherwise, classify the cause as an external oscillatory disturbance.
- Integrated Diagnosis: Synthesize the results from steps 3, 4, and 5 to issue a final diagnostic report stating the identified single or multiple root causes.
- 4.0 Data Analysis: The final output is a qualitative diagnosis of the root cause(s). The Hammerstein model provides a binary output for stiction, while the amplitude discrimination algorithm classifies the oscillation as originating from tuning or disturbance.
Protocol: Performance Evaluation of Routing Protocols in Smart Grid Environments
This protocol outlines a method to evaluate network routing protocols, the oscillation or divergence of which can impact DOS convergence in smart grid communications [53].
- 1.0 Objective: To evaluate and compare the performance of different multi-hop routing protocols in harsh smart grid spectrum environments to establish design guidelines.
- 2.0 Prerequisites:
- Network simulator (e.g., ns-2).
- Implementation of routing protocols (e.g., AODV, DYMO, DSDV, TUQR).
- Smart grid environmental characteristics data (e.g., from real-world field tests).
- 3.0 Procedure:
- Simulation Setup: Configure the network simulator with smart grid environmental characteristics, including background noise, attenuation, and wireless propagation phenomena modeled on real-world data from locations like a 500 kV outdoor substation or an underground network transformer vault.
- Protocol Implementation: Select and configure the routing protocols for testing. This typically includes on-demand (AODV, DYMO), table-driven (DSDV), and QoS-aware (TUQR) protocols.
- Define Metrics: Set up the simulation to measure Packet Delivery Ratio (PDR), end-to-end delay, and network energy consumption.
- Run Experiments: Execute multiple simulation runs for each protocol in each defined smart grid environment to gather statistically significant data.
- Performance Comparison: Analyze the results to compare the performance of the different protocols against the defined metrics.
- 4.0 Data Analysis: Quantitative analysis of PDR (higher is better), end-to-end delay (lower is better), and energy consumption (lower is better). Results are used to formulate routing design guidelines for smart grid applications, such as preferring QoS-aware or adaptive protocols [53].
Diagnostic Workflow and Signaling Pathways
The following diagram illustrates the integrated logical workflow for diagnosing the root cause of oscillations in a control system, as detailed in Section 3.1.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Computational and Analytical Tools for Grid Oscillation Research This table details key software, algorithms, and data types required for conducting research in grid oscillation and DOS convergence.
| Tool / Reagent | Type | Function in Research |
|---|---|---|
| Hiking Optimization Algorithm (HOA) | Metaheuristic Algorithm | Optimizes complex grid objectives (cost, losses, voltage) by leveraging an adaptive search mechanism to avoid local optima [54]. |
| Hammerstein Model | Identification Model | Detects and quantifies the presence of control valve stiction, a common nonlinearity causing oscillations, from PV and OP data [52]. |
| ns-2 Network Simulator | Simulation Platform | Evaluates the performance of communication and routing protocols under harsh smart grid conditions before real-world deployment [53]. |
| Hilbert-Huang (HH) Spectrum | Signal Processing Technique | Used in amplitude-based discrimination algorithms to analyze the time-frequency characteristics of oscillations for root cause classification [52]. |
| Process Variable (PV) & Controller Output (OP) Data | Time-Series Data | The primary dataset for model-based diagnosis; used as input for stiction detection and oscillation characterization algorithms [52]. |
In the broader context of research focused on setting energy grid parameters for Density of States (DOS) convergence, achieving a stable and converged Self-Consistent Field (SCF) calculation is a critical prerequisite. The accuracy of the resulting DOS is entirely dependent on the quality of the underlying converged wavefunction. Many challenging systems, such as open-shell transition metal complexes, metallic surfaces, and slabs, are prone to SCF convergence difficulties, manifesting as oscillatory behavior or a complete failure to reach the designated convergence criteria [55] [56]. Among the various strategies available, adopting conservative mixing parameters serves as a fundamental technique for stabilizing the SCF procedure. This application note details the systematic application of decreased SCF%Mixing and DIIS%Dimix settings, providing robust protocols for researchers and development professionals to overcome persistent convergence barriers.
Theoretical Background: The Role of Mixing and DIIS
The SCF procedure iteratively solves the Kohn-Sham equations by generating a new electron density from the output orbitals of the previous cycle. Density mixing is the process where a fraction of this new output density is mixed with the density from previous cycles to construct the input for the next iteration. A high mixing parameter (e.g., 0.3) leads to large steps between iterations, which can speed up convergence in simple systems but often induces oscillations in difficult cases.
The Direct Inversion in the Iterative Subspace (DIIS) method accelerates convergence by constructing a new trial density from a linear combination of densities from several previous iterations [55]. The DIIS%Dimix parameter controls the aggressiveness of this extrapolation. Conservative tuning involves reducing both the SCF%Mixing and DIIS%Dimix values, thereby taking smaller, more stable steps toward the solution at the cost of a potentially higher number of SCF cycles.
Parameter Specifications and Defaults
The table below summarizes the default, aggressive, and recommended conservative values for key SCF parameters, drawing from established documentation [55].
Table 1: SCF Convergence Parameter Comparison
| Parameter | Typical Default / Aggressive Value | Recommended Conservative Value | Function |
|---|---|---|---|
SCF%Mixing |
0.1 - 0.3 | 0.05 [55] | Fraction of new density mixed into the input for the next cycle. |
DIIS%Dimix |
Not Specified (Larger) | 0.1 [55] | Controls the aggressiveness of the DIIS extrapolation. |
DIIS%Adaptable |
True |
False [55] |
Disables automatic adjustment of Dimix for a fixed, predictable strategy. |
Integrated Protocol for Conservative Tuning and DOS Convergence
This section provides a detailed, step-by-step workflow for implementing conservative SCF parameters, with a specific focus on ensuring the resulting wavefunction is suitable for high-quality DOS calculations.
Preliminary Analysis and System Assessment
- System Characterization: Before beginning, identify features of your system that make convergence challenging. These commonly include:
- Initial Low-Level Calculation: For a system with a suspected difficult convergence, start with a smaller basis set (e.g., SZ) to generate an initial density and wavefunction guess [55]. This converged result can then be used as a restart for a calculation with the target larger basis set.
- Check for Linear Dependency: If a calculation aborts due to a dependent basis error, address this first by applying confinement to diffuse basis functions or removing them, as conservative mixing will not resolve this fundamental issue [55].
Core Protocol: Implementing Conservative Parameters
The following input block demonstrates the direct implementation of conservative parameters in a typical computational input file.
Code Block 1: Example input snippet for applying conservative SCF parameters.
Protocol Steps:
- Initial Application: Introduce the parameters from Code Block 1 into your input file after an initial SCF failure or upon observing oscillatory behavior in the SCF energy.
- Monitor Convergence: Run the calculation and carefully monitor the output log for the change in total energy and density between cycles. The convergence should become smoother and more monotonic.
- Iterative Refinement: If convergence is still not achieved, consider further reducing
SCF%Mixingto 0.02 or 0.01. Alternatively, switch to a more robust SCF algorithm like the MultiSecant method [55], which can be invoked withSCF; Method MultiSecant; End. - Final Convergence for DOS: Once the SCF has converged to a tight criterion (e.g.,
!TightSCFin ORCA [56] [57]), proceed with the DOS calculation. Ensure theDOS%DeltaEparameter is set to a sufficiently small value (e.g., 0.01 eV) to obtain a smooth density of states, and verify that the k-space grid is well-converged.
Workflow Visualization
The following diagram illustrates the logical decision process and workflow for applying conservative tuning within a larger DOS convergence project.
Diagram Title: SCF Troubleshooting and DOS Workflow
The Scientist's Toolkit: Essential Research Reagents
The following table lists key "reagents" or computational tools referenced in this protocol.
Table 2: Key Research Reagent Solutions
| Item / Keyword | Function / Description | Context of Use |
|---|---|---|
| Conservative Mixing | Stabilizes SCF cycles by taking smaller steps in density update. | Primary remedy for oscillating or diverging SCF calculations. |
| MultiSecant Method | An alternative SCF convergence algorithm that can be more robust than DIIS [55]. | Used when conservative DIIS parameters fail. |
| NumericalQuality Good | Improves the quality of the numerical integration grid and density fitting [55]. | Addresses convergence issues stemming from numerical inaccuracies. |
| EngineAutomations | Allows SCF parameters (e.g., electronic temperature) to change during a geometry optimization [55]. | Used in geometry optimizations where initial forces are large. |
| SZ Basis Set | A small, minimal basis set. | Generating an initial wavefunction guess for difficult systems. |
Concluding Remarks
The strategic decrease of SCF%Mixing and DIIS%Dimix parameters is a proven, conservative approach to taming difficult SCF calculations. While it may increase the time to convergence, its reliability in producing a stable wavefunction is unparalleled for problematic systems. This stable SCF result is the essential foundation upon which all subsequent analysis, including accurate and converged Density of States, is built. Integrating this protocol into the early stages of energy grid parameter research ensures that DOS convergence studies are conducted on a reliable electronic ground state.
In the pursuit of accurate electronic structure calculations for materials critical to energy grid research, such as those used in conduction bands or battery materials, achieving a well-converged Density of States (DOS) is a fundamental objective. A significant obstacle on this path is the emergence of linear dependency within the basis set, a numerical instability that can compromise the integrity of the entire calculation. This occurs when the set of basis functions used to describe the electron orbitals ceases to be linearly independent, often due to the inclusion of overly diffuse functions in systems with high coordination or periodic boundary conditions [55]. Such dependencies render the overlap matrix singular or nearly singular, jeopardizing the numerical accuracy of results [55].
This Application Note provides a detailed protocol for identifying and resolving basis set linear dependency, with a particular emphasis on the use of confinement potentials as a primary mitigation strategy. The guidance is framed within the context of optimizing grid parameters for DOS convergence, a crucial step for predicting electronic properties in energy materials.
Background and Core Concepts
The Linear Dependency Problem
In linear combination of atomic orbitals (LCAO) calculations, the program constructs Bloch functions from the elementary basis functions for each k-point in the Brillouin Zone (BZ). The overlap matrix of this Bloch basis is then diagonalized. A finding that the smallest eigenvalue is zero indicates a linearly dependent basis. Given the finite precision of numerical computations, trouble arises even before this exact point is reached; if the smallest eigenvalue is very small, the basis is considered numerically unstable [55].
The root cause is often the diffuseness of certain basis functions, especially in highly coordinated atoms or slab systems [55]. These diffuse functions exhibit significant overlap with many neighboring atoms, leading to a loss of numerical independence. In the context of DOS convergence for energy grid materials, an unconverged or unstable basis set will produce spurious peaks or incorrect band gaps, directly impacting the reliability of subsequent property predictions.
The Confinement Solution
Confinement addresses this issue by systematically reducing the spatial extent of diffuse basis functions. By applying a confining potential, the tail of the orbital is forced to decay more rapidly, thereby minimizing excessive overlap with distant neighbors and restoring linear independence. The Confinement key in software packages like BAND allows users to control this process [55]. A strategic approach involves applying confinement selectively; for instance, in a slab calculation, one might use a normal basis for surface atoms (to properly describe decay into vacuum) and a confined basis for inner slab atoms where such diffuseness is unnecessary [55].
Experimental Protocols
Protocol 1: Diagnosing Linear Dependency
Objective: To identify and confirm the presence of linear dependency in a basis set. System: Bulk semiconductor (e.g., Silicon) or metallic slab (e.g., Palladium).
- Calculation Setup: Begin a standard single-point energy calculation with a high-quality, potentially diffuse, basis set (e.g., a Triple-Zeta Polarized basis).
- Error Monitoring: Execute the calculation. A controlled termination of the job with an explicit error message about a "dependent basis" is a positive indicator of the issue.
- Overlap Matrix Analysis: In the output log, locate the results of the overlap matrix diagonalization for each k-point.
- Eigenvalue Inspection: Identify the smallest eigenvalue of the overlap matrix. The program will compare this value against a pre-defined
Dependencycriterion (configurable via theBaskeyword). A smallest eigenvalue significantly below this criterion (e.g., < 1e-7) confirms numerical linear dependency [55].- Note: It is strongly advised not to simply adjust the
Dependencycriterion to bypass the error, as this compromises numerical accuracy [55].
- Note: It is strongly advised not to simply adjust the
Protocol 2: Resolving Dependency via Orbital Confinement
Objective: To eliminate linear dependency by applying a radial confinement potential to basis orbitals.
- Identify Diffuse Orbitals: From the diagnostic output, determine which atomic species and angular momentum channels (e.g.,
d-orbitals in a transition metal) are contributing to the small eigenvalues. - Define Confinement Potential: In the basis set definition for the problematic atom, apply a confining potential. The following table summarizes the key parameters for a
ConfinedOrbitalas used in QuantumATK [60]:- Radial Cutoff Radius (
radial_cutoff_radius): The hard cutoff limit for the orbital. - Confinement Start Radius (
confinement_start_radius): The radius at which the confining potential begins to act. - Confinement Strength (
confinement_strength): The magnitude of the confining potential, typically in units of Hartree*Bohr [60].
- Radial Cutoff Radius (
- Parameter Selection Strategy:
- Start with a
confinement_start_radiusset to 0.7-0.8 times the originalradial_cutoff_radiusof the unconfined orbital. - Use a
confinement_strengthof 20.000 * Hartree * Bohr as an initial value [60]. - The
radial_step_sizefor numerically generating the orbital can often be left at a default value of 0.001*Bohr [60].
- Start with a
- Iterative Re-testing: Run the calculation again with the confined basis set. Monitor the smallest eigenvalue of the overlap matrix to ensure it has risen above the dependency threshold. Iteratively adjust the confinement parameters if necessary.
Protocol 3: Strategic Removal of Basis Functions
Objective: To resolve linear dependency by manually removing the most diffuse basis functions from the set.
- Basis Set Analysis: Examine the composition of your basis set. Identify the most diffuse function(s) based on their exponent values (the smallest exponents correspond to the most diffuse functions).
- Create a Truncated Set: Generate a new, smaller basis set by removing one or more of these diffuse functions. This is often a last resort, as it can reduce the basis set quality and flexibility.
- Validation: Run the calculation with the truncated basis set and check for:
- Elimination of Error: The dependency error should be absent.
- DOS Quality: Compare the resulting DOS with a reference (e.g., from a planewave calculation). Ensure that the key features, especially near the Fermi level, are preserved and that the DOS is smooth and physically reasonable. The goal is a converged DOS where adding back the diffuse function does not significantly change its character.
The following workflow diagram illustrates the decision process for addressing linear dependency:
Data Presentation and Analysis
Quantitative Impact of Confinement on DOS Convergence
The table below summarizes the expected effects of different confinement strategies on key calculation metrics, including the DOS convergence quality.
Table 1: Comparison of Basis Set Optimization Strategies for DOS Convergence
| Strategy | Basis Set Size | Overlap Matrix Min. Eigenvalue | Total Energy Change (per atom) | Band Gap Error | DOS Convergence Quality |
|---|---|---|---|---|---|
| Original (Diffuse) | Large | < 1.0e-7 (Fails) | N/A | N/A | Unreliable |
| Medium Confinement | Unchanged | 1.0e-4 (Stable) | < 0.7 mEh [61] | < 20 meV [61] | Adequate |
| Strong Confinement | Unchanged | 1.0e-2 (Very Stable) | 1-5 mEh | 50-100 meV | Potentially Deteriorated |
| Function Removal | Reduced | > 1.0e-4 (Stable) | 5-10 mEh | 50-200 meV | Requires Validation |
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Computational Tools for Basis Set Optimization
| Item / Keyword | Function | Example Usage / Note |
|---|---|---|
Confinement Key |
Applies a radial potential to reduce orbital diffuseness, directly combating linear dependency [55]. | Use selectively on inner atoms in slabs. |
ConfinedOrbital |
A specific orbital type whose range is controlled by parameters like radial_cutoff_radius and confinement_strength [60]. |
confinement_strength=20.000*Hartree*Bohr [60]. |
Dependency Criterion |
The threshold (set via Bas keyword) for the smallest eigenvalue of the overlap matrix [55]. |
Do not relax to bypass errors without fixing the root cause. |
GramSchmidtOrthonormalization |
A boolean parameter that can be set to True to automatically transform basis orbitals into an orthonormal set [60]. |
Helps ensure numerical stability from the start. |
| Uncontracted Basis Sets | Basis sets using only primitive Gaussian-type orbitals (GTOs), which can help avoid linear dependency inherent in some contracted sets for solids [61]. | e.g., unc-def2-QZVP-GTH [61]. |
| Overlap Matrix Analysis | The primary diagnostic tool. Diagonalizing this matrix reveals the eigenvalues that indicate linear dependency [55]. | The key output for Protocol 1. |
Integrated Workflow for Energy Grid Parameter Research
For research focused on setting energy grid parameters for reliable DOS convergence, the management of basis set quality is a non-negotiable prerequisite. The following diagram integrates the protocols above into a comprehensive workflow for a materials study.
This workflow ensures that the foundation of your calculation—the basis set—is robust and numerically stable before you invest resources in the final DOS analysis and the extraction of electronic properties for energy grid materials.
In computational chemistry and materials science, the precision of property prediction is fundamentally tied to the numerical methods employed for integration and density fitting. This application note details protocols for enhancing numerical accuracy, with a specific focus on configuring integration grids and density fitting approximations. The procedures are framed within the critical context of achieving converged and reliable Density of States (DOS) calculations, a cornerstone for electronic structure analysis in drug development and materials research. Accurate DOS profiles are indispensable for understanding reactivity, bonding, and electronic transitions, making the underlying numerical robustness a primary concern for scientific investigation.
Theoretical Background and Key Concepts
Numerical Integration in Density Functional Theory
In Density Functional Theory (DFT) calculations, the exchange-correlation energy is evaluated through numerical integration, as an analytical solution is typically intractable. The default numerical integration grid in many software packages, such as ADF, is a refined version of the Becke grid [62]. This scheme partitions molecular space into atomic-centered regions using a fuzzy cell method, and the integration accuracy is controlled by the grid's quality. The implementation employs a partition function that depends on the distance from the atoms and element-specific parameters [62].
For a given grid quality, the number of radial shells and angular points directly determines the computational cost and accuracy. Pruned grids, which use a non-uniform number of angular points across different radial shells, are optimized to provide a specific accuracy level with a minimal number of points [63] [64].
Density Fitting for Computational Efficiency
Density fitting (DF), also known as the resolution of the identity (RI) approximation, is a technique that significantly accelerates the computation of two-electron Coulomb integrals in Hartree-Fock and DFT calculations [65]. By approximaching the electron density with an auxiliary basis set, it reduces the formal scaling of the computation. Molpro documentation emphasizes that density fitting is "highly recommended... as the induced errors are negligible and it offers massive speed increases, particularly for pure functionals" [65]. This makes it an essential tool for large systems, such as those encountered in drug development.
Quantitative Data on Integration Grids
The choice of integration grid has a direct, quantifiable impact on numerical accuracy. The following tables summarize standard grid specifications and their associated accuracies across different computational software packages.
Table 1: Standard Integration Grid Specifications and Their Accuracies
| Grid Name (Quality) | Radial Shells (mmm) | Angular Points (nnn) | Total Points per Atom (approx.) | Typical Use Case & Software Context |
|---|---|---|---|---|
| CoarseGrid | 35 | 110 | 3,850 | Initial scans, testing; pruned grid [64]. |
| SG1Grid | 50 | 194 | 9,700 | Obsolete; not recommended for production [63] [64]. |
| Normal (Default) | Not Specified | Not Specified | ~Equivalent to INTEGRATION 4 (ADF) [62] | Standard production calculations; default in ADF [62]. |
| FineGrid | 75 | 302 | ~7,000 | Default pruned grid in some programs; good for production [63] [64]. |
| Good | Not Specified | Not Specified | ~Equivalent to INTEGRATION 6 (ADF) [62] | Higher accuracy calculations [62]. |
| UltraFine | 99 | 590 | ~58,000 | Molecules with tetrahedral centers; very low frequency modes [63] [64]. |
| SuperFineGrid | 150/225* | 974 | ~150,000-220,000 | Very high accuracy requirements [64]. |
*150 for first two periodic table rows, 225 for later elements [64].
Table 2: Software-Specific Grid Control Parameters and Defaults
| Software | Primary Grid Control Keyword | Key Accuracy Parameter | Default Value | Key Functional Considerations |
|---|---|---|---|---|
| ADF | BECKEGRID |
Quality [Basic...Excellent] |
Normal [62] |
Automatically boosts radial grids for sensitive meta-GGAs/Hybrids [62]. |
| Molpro | GRID (on KS command) |
GRIDTHR=target |
1.d-6 (per atom) [65] |
Tighter grids recommended for meta-GGA functionals [65]. |
| Gaussian | Integral(Grid=GridName) |
Grid name (e.g., FineGrid) |
FineGrid [64] |
CPHF calculations use a coarser grid by default [64]. |
Experimental Protocols for DOS Convergence
Protocol 1: Benchmarking Integration Grid for DOS Convergence
This protocol provides a systematic method for determining the integration grid necessary for a numerically-converged DOS calculation for a given system and functional.
1. Problem Definition and Initial Setup
- Objective: To establish the minimum integration grid quality that yields a converged DOS for subsequent research.
- System Preparation: Generate a well-defined, optimized molecular or crystalline structure.
- Software and Method Selection: Choose a consistent computational package (e.g., ADF, Molpro, Gaussian), DFT functional, and a high-quality basis set. The DOS method (e.g., with a specific broadening width) must be fixed throughout the benchmark.
2. Hierarchical Grid Testing
- Begin calculations with a CoarseGrid or
Quality=Basicsetting. - Progressively increase the grid quality through the hierarchy (e.g.,
Normal->Good->VeryGood->UltraFine). - For each grid level, perform a single-point energy and property calculation to generate the DOS.
3. Convergence Analysis
- Primary Metric: Visually compare the DOS spectra, paying close attention to the position and shape of the valence band maximum, conduction band minimum, and any key frontier molecular orbitals or peak structures.
- Quantitative Metric: Calculate the root-mean-square deviation (RMSD) of the DOS between successive grid quality levels. Convergence is achieved when the RMSD falls below a predefined threshold (e.g., 1-5 meV for energy levels or 1% for relative peak intensities).
- Secondary Check: Monitor the total energy change. While DOS convergence is often achieved before total energy convergence, a large energy shift may indicate an inadequate grid.
4. Result Documentation and Selection
- Document the RMSD values and visual comparisons for each grid level.
- The final recommended grid is the least computationally expensive one that meets the convergence criteria.
The following workflow diagram illustrates the hierarchical benchmarking process:
Protocol 2: Validating Density Fitting for DOS Calculations
This protocol ensures that the use of density fitting does not introduce significant error into the DOS compared to a conventional calculation.
1. Problem Definition
- Objective: To validate that a chosen auxiliary basis set for density fitting reproduces the conventional DOS within an acceptable error margin.
2. Reference Calculation
- Perform a conventional (non-density-fitted) calculation using the integration grid established in Protocol 1. This serves as the reference DOS.
3. Density-Fitted Calculations
- Run a series of calculations with density fitting enabled, using different auxiliary basis sets. Typically, one would test auxiliary sets of increasing size and quality that are designed for the primary basis set being used.
4. Validation and Error Analysis
- Primary Metric: Compare the DOS from the DF calculations to the reference DOS.
- Quantitative Metric: Calculate the RMSD of the DOS for each DF calculation against the reference.
- Acceptance Criterion: A suitable auxiliary basis set is one where the RMSD of the DOS is below the predefined threshold (e.g., comparable to the grid convergence threshold). The total energy difference should also be examined.
5. Performance Assessment
- Document the computational time and memory usage for the validated DF calculation compared to the conventional one. This highlights the efficiency gain.
Protocol 3: Combined Grid and Fitting Optimization for Sensitive Functionals
Meta-GGA and hybrid functionals are more sensitive to numerical quadrature. This protocol combines the first two protocols with specific adjustments for such functionals.
1. Initial Setup with Defaults
- Select the sensitive functional (e.g., M06-L, M06-2X, SCAN) [62].
- Note that some software, like ADF, may automatically apply a
RadialGridBoostfor known sensitive functionals [62]. In Molpro, it is recommended to "use tighter grid thresholds than those set by default... if meta-GGA type functionals are used" [65].
2. Grid Refinement Loop
- Follow Protocol 1, but start from a higher baseline grid (e.g.,
GoodorFineGrid). - Pay particular attention to the core-valence and low-energy conduction regions in the DOS, which can be more severely affected by numerical noise from these functionals.
3. Density Fitting Validation
- Execute Protocol 2 using the refined grid from the previous step. For hybrid functionals, ensure the exchange integral evaluation is compatible with the density fitting approach.
4. Final Recommendation
- The output is a numerically stable and efficient configuration (functional + grid + density fitting basis) for obtaining a reliable DOS.
The Scientist's Toolkit: Research Reagent Solutions
This section lists essential "research reagents" – the software commands, parameters, and basis sets – required to perform the experiments described in the protocols.
Table 3: Essential Computational Tools for Numerical Accuracy Enhancement
| Item Name | Function / Purpose | Example / Specification |
|---|---|---|
| Becke Grid Quality Keywords | Controls the fineness of the molecular integration grid in ADF [62]. | Basic, Normal, Good, VeryGood, Excellent |
| Predefined Grid Keywords | Specifies standard integration grids in Gaussian & other codes [63] [64]. | CoarseGrid, FineGrid, UltraFineGrid, SuperFineGrid |
| Custom Grid Specification | Allows for manual, fine-grained control over the grid structure [63]. | Grid=99302 for (99 radial, 302 angular) points |
| Auxiliary Basis Sets | The "density fitting basis" used to approximate electron density, critical for accuracy & speed [65]. | Examples: def2-QZVP/JKFIT, cc-pVTZ-RI |
| Radial Grid Boost | Manually increases radial points for sensitive functionals if not automatic [62]. | ADF: RadialGridBoost [factor] |
| Grid Accuracy Threshold | Sets a target accuracy for automatic grid generation in Molpro [65]. | GRIDTHR=1.d-7 (Tighter than default) |
| Density Fitting Command Prefix | Invokes the density fitting approximation in Molpro [65]. | DF-RKS (For restricted KS-DFT) |
Workflow Integration Diagram
The following diagram summarizes the logical relationship and integration of the three protocols into a comprehensive workflow for securing numerical accuracy in DOS calculations, particularly relevant for the demanding context of drug development research.
This Application Note details a structured, multi-level workflow for performing sequential convergence studies of Density of States (DOS) from the minimal SZ basis set to larger, more accurate basis sets. Accurately calculating the DOS is fundamental to understanding the electronic properties of materials, which in turn is critical for designing novel compounds in pharmaceutical development and materials science. The protocol is explicitly framed within broader research aimed at setting robust energy grid parameters to ensure the convergence and reliability of DOS calculations. By providing a standardized yet flexible framework, this document serves researchers, scientists, and drug development professionals in streamlining their computational experiments, enhancing reproducibility, and accelerating the discovery pipeline.
The proposed methodology is built upon a formal abstraction hierarchy, adapting proven concepts from biofoundries and scientific computing to the domain of quantum chemistry [66]. This hierarchy organizes the complex computational experiment into four distinct, interoperable levels, ensuring clarity, modularity, and reusability.
- Level 0: Project: This is the overarching goal: "Perform a Sequential DOS Convergence Study from SZ to a larger basis set (e.g., TZ) for a target molecule."
- Level 1: Service/Capability: This level defines the high-level functions required to complete the project. Examples include "Geometry Optimization Service," "Single Point Energy Calculation Service," and "DOS Convergence Analysis Service."
- Level 2: Workflow: Each service is decomposed into modular, sequential workflows corresponding to the Design-Build-Test-Learn (DBTL) cycle [66]. A workflow is a sequence of tasks that delivers a specific function, such as "Molecular Structure Preparation" (Design) or "Basis Set Convergence Testing" (Learn).
- Level 3: Unit Operation: This is the most fundamental level, comprising individual, automated computational tasks performed by specific software or scripts. Examples include "File Format Conversion," "Quantum Chemistry Job Submission," or "Data Extraction from Log File."
This hierarchical structure allows researchers to operate at the appropriate level of abstraction, enabling the combination and re-use of atomic workflows to construct complex meta-workflows for sophisticated scientific experiments [67].
Table: Abstraction Hierarchy for DOS Convergence Studies
| Level | Name | Description | Example in DOS Convergence |
|---|---|---|---|
| 0 | Project | The overall scientific goal or experiment. | Achieve converged DOS for Drug Candidate X. |
| 1 | Service/Capability | A high-level function provided to fulfill the project. | DOS Convergence Analysis Service. |
| 2 | Workflow | A DBTL-stage-specific sequence of tasks. | Basis Set Convergence Testing (Learn). |
| 3 | Unit Operation | An individual task performed by hardware/software. | Execute Gaussian SP calculation. |
Application Notes & Experimental Protocols
Meta-Workflow Orchestration
The sequential convergence study is implemented as a meta-workflow, a complex workflow orchestrated from several self-contained, atomic sub-workflows [67]. This approach promotes sharing, reusability, and modular testing of individual workflow components. The entire process is formally described by an abstract workflow (defining the structure and data flow), a concrete workflow (an instance for a specific computational engine like Gaussian), a workflow configuration (parameters and input files), and the workflow engine itself [67].
Detailed Experimental Protocols
Protocol 1: Molecular System Preparation (Design Workflow)
Objective: To prepare and validate the initial molecular geometry for all subsequent quantum chemical calculations.
- Acquire Initial Structure: Obtain the 3D molecular structure from a database (e.g., PubChem) or generate it using chemical drawing software.
- Geometry Optimization Preprocessing:
a. Input File Generation (Unit Operation): Create a Gaussian input file (
.comor.gjf) with the following specifications: b. Coordinate System Check: Ensure the use of Cartesian coordinates for maximum compatibility. - Submit Optimization Job (Unit Operation): Execute the input file on the available computing infrastructure.
- Validate Optimized Geometry (Unit Operation):
- Confirm the job terminated normally.
- Verify the structure resides at a true minimum by checking for the absence of imaginary frequencies in the vibrational analysis.
Protocol 2: Single Point Energy & DOS Calculation (Build-Test Workflow)
Objective: To compute the single point energy and Density of States for the optimized geometry using a sequence of basis sets.
- Basis Set Sequence Definition: Define the ordered list of basis sets for the convergence study. Example sequence:
SZ < DZ < TZ < QZ. - Iterative Single Point Calculation: a. Input File Generation (Unit Operation): For each basis set in the sequence, generate a Gaussian input file for the pre-optimized geometry requesting a DOS calculation. b. Job Submission & Monitoring (Unit Operation): Submit each single point job and monitor for successful completion.
- Data Extraction (Unit Operation): Upon successful completion of each job, parse the output log file to extract key quantitative data, including:
- Total Single Point Energy (Hartrees)
- HOMO-LUMO Gap (eV)
- Fermi Energy Level (eV)
Protocol 3: Convergence Analysis (Learn Workflow)
Objective: To determine the basis set at which the DOS and related electronic properties are converged within a defined threshold.
- Data Aggregation (Unit Operation): Compile all extracted data into a structured table.
- Delta (Δ) Calculation (Unit Operation): For each property (Energy, HOMO-LUMO Gap), calculate the difference relative to the calculation with the largest basis set (e.g., QZ).
ΔEnergy = |Energy_BasisSet - Energy_QZ| - Convergence Threshold Check: Compare the calculated Δ values against a predefined convergence threshold (e.g., ΔEnergy < 1.0 kcal/mol). The basis set where the Δ values fall and remain below the threshold for all subsequent calculations is identified as the converged basis set.
Data Presentation
The following tables summarize the quantitative data extracted from a hypothetical DOS convergence study for a model compound, following the protocols outlined above.
Table: Extracted Single Point Calculation Data
| Basis Set | Total Energy (Hartree) | HOMO-LUMO Gap (eV) | Fermi Level (eV) |
|---|---|---|---|
| SZ | -405.7621 | 4.85 | -2.10 |
| DZ | -406.8915 | 5.12 | -2.25 |
| TZ | -406.9238 | 5.18 | -2.28 |
| QZ | -406.9280 | 5.19 | -2.29 |
Table: Convergence Analysis (Δ relative to QZ)
| Basis Set | Δ Energy (kcal/mol) | Δ HOMO-LUMO Gap (eV) | Converged? (Y/N) |
|---|---|---|---|
| SZ | 104.0 | 0.34 | N |
| DZ | 22.9 | 0.07 | N |
| TZ | 2.6 | 0.01 | Y (Within Threshold) |
| QZ | 0.0 | 0.00 | Y |
Workflow Visualization
The following diagram illustrates the logical flow and data dependencies of the meta-workflow for the sequential DOS convergence study, from project initiation to the final analysis.
Diagram: Multi-level Meta-workflow for DOS Convergence
The Scientist's Toolkit: Research Reagent Solutions
This section details the essential computational "reagents" and materials required to execute the described DOS convergence meta-workflow.
Table: Essential Research Reagents & Computational Materials
| Item Name | Function / Purpose | Example / Specification |
|---|---|---|
| Quantum Chemistry Software | Performs the core electronic structure calculations, including geometry optimization, single point energy, and DOS computation. | Gaussian 16, ORCA, GAMESS. |
| Basis Set Library | A predefined collection of mathematical functions (basis sets) used to represent molecular orbitals. The central object of the convergence study. | Pople-style (e.g., 6-31G*), Dunning-style (e.g., cc-pVTZ), or minimal (e.g., SZ). |
| Molecular Structure File | The initial 3D coordinate data of the molecule under investigation. | .mol, .sdf, or .xyz file format. |
| Computational Job Scheduler | Manages the submission and execution of computational jobs on high-performance computing (HPC) clusters. | Slurm, PBS Pro. |
| Data Parsing Script | An automated script (e.g., in Python) to extract key quantitative data (energies, orbital levels) from bulky output files. | Custom Python script using cclib library. |
| Visualization & Analysis Tool | Software used to plot results, analyze convergence trends, and visualize the Density of States. | Origin, Matplotlib, GaussView. |
Within the broader scope of research focused on setting energy grid parameters for Density of States (DOS) convergence, the initial preparation of molecular structures is a critical, yet often overlooked, prerequisite. A successfully converged geometry optimization provides the foundational atomic coordinates upon which all subsequent electronic structure analyses, including DOS calculations, depend. An unconverged or poorly optimized geometry can lead to inaccurate electronic energies, faulty force calculations, and ultimately, a non-representative DOS, compromising the entire research effort. This document outlines application notes and protocols to ensure molecular structures are robustly prepared for the convergence process, thereby enhancing the reliability and reproducibility of results in scientific and drug development research.
Core Concepts and Quantitative Convergence Criteria
Geometry optimization is an iterative process that adjusts a system's nuclear coordinates to locate a local minimum on the potential energy surface (PES), moving "downhill" in energy until the structure is converged [68]. Convergence is typically monitored through a combination of criteria related to energy changes, forces (gradients), and the step size between iterations.
The strictness of these criteria can be quickly set using predefined quality levels, which scale all thresholds simultaneously [68]. The following table summarizes these standard settings:
Table 1: Standard convergence quality levels and their associated thresholds [68].
| Quality Setting | Energy (Ha) | Gradients (Ha/Å) | Step (Å) | StressEnergyPerAtom (Ha) |
|---|---|---|---|---|
| VeryBasic | 10⁻³ | 10⁻¹ | 1 | 5×10⁻² |
| Basic | 10⁻⁴ | 10⁻² | 0.1 | 5×10⁻³ |
| Normal (Default) | 10⁻⁵ | 10⁻³ | 0.01 | 5×10⁻⁴ |
| Good | 10⁻⁶ | 10⁻⁴ | 0.001 | 5×10⁻⁵ |
| VeryGood | 10⁻⁷ | 10⁻⁵ | 0.0001 | 5×10⁻⁶ |
A geometry optimization is considered converged only when all the following conditions are met [68]:
- The energy change between steps is smaller than the
Convergence%Energythreshold multiplied by the number of atoms. - The maximum Cartesian nuclear gradient is smaller than the
Convergence%Gradientsthreshold. - The root mean square (RMS) of the Cartesian nuclear gradients is smaller than 2/3 of the
Convergence%Gradientsthreshold. - The maximum Cartesian step is smaller than the
Convergence%Stepthreshold. - The root mean square (RMS) of the Cartesian steps is smaller than 2/3 of the
Convergence%Stepthreshold.
It is important to note that the step threshold is a less reliable measure of coordinate precision than the gradients. For accurate results, it is recommended to tighten the gradient criterion rather than the step criterion [68].
Workflow and Decision Pathway for Robust Pre-optimization
The following diagram outlines a systematic protocol for preparing a molecular system for geometry optimization, from initial setup to troubleshooting a converged structure. This workflow is designed to prevent common pitfalls and ensure the resulting geometry is a true local minimum suitable for DOS calculations.
Title: Decision pathway for geometry pre-optimization and troubleshooting.
Detailed Experimental Protocols
Protocol A: Standard Pre-optimization for Stable Molecules
This protocol is suitable for most small to medium-sized, well-behaved organic molecules in the gas phase or solution.
- Initial Structure Preparation: Generate a reasonable 3D structure using a molecular builder or from a crystal structure database. Ensure proper bond connectivity and stereochemistry.
- Software and Method Selection:
- Software: AMS, Gaussian, ORCA, or other quantum chemistry packages.
- Method: Select an appropriate level of theory (e.g., DFT with a functional like B3LYP and a basis set like 6-31G*).
- Optimization Parameters:
- Coordinate System: Use redundant internal coordinates (
opt=Redundantin Gaussian) for most molecular systems, as they often lead to faster convergence [69]. - Initial Hessian: For systems with "normal" bonding, the default approximate Hessian is sufficient.
- Convergence Criteria: Start with the
Normalquality setting (see Table 1). - Max Iterations: Use the default value, which is typically large enough for most systems.
- Coordinate System: Use redundant internal coordinates (
- Execution and Monitoring: Run the optimization and monitor the output for steady, downhill progress in energy. The job is successful when all convergence criteria are met.
- Post-Optimization Verification: Confirm the optimized structure is a minimum by performing a frequency calculation (a.k.a. Hessian calculation) to ensure there are no imaginary frequencies.
Protocol B: Tight Optimization for DOS-Grade Structures
This protocol is for obtaining high-precision geometries required as input for subsequent DOS convergence studies. It uses stricter thresholds and is more computationally demanding.
- Initial Structure: Use a pre-optimized structure from Protocol A as the starting point.
- Software and Method Selection: Use the same method and basis set as the initial optimization to ensure consistency.
- Optimization Parameters:
- Convergence Criteria: Use the
GoodorVeryGoodquality setting. This tightens the gradient criterion, which is key for accurate final coordinates [68]. - Initial Hessian: For the final, tight optimization, it is recommended to calculate the initial Hessian explicitly (
opt=calcfcin Gaussian) to provide the optimizer with an accurate starting point for the energy landscape [69].
- Convergence Criteria: Use the
- Execution: Run the optimization. This will require more computational time and SCF cycles per point due to the need for more accurate and noise-free gradients.
- Verification: A final frequency calculation is mandatory to unequivocally confirm the structure is a minimum.
Protocol C: Troubleshooting a Stalled or Oscillating Optimization
This protocol is activated when an optimization fails to converge within the maximum number of cycles or shows oscillatory behavior.
- Diagnosis: Inspect the optimization output. Check if the energy, gradient, or step values are oscillating without a clear downward trend.
- Initial Hessian Intervention: The most common remedy is to provide a better initial Hessian. Restart the optimization from the last (or best) geometry with the
opt=calcfcoption [69]. - Coordinate System Change: If the problem persists, switch the coordinate system. For example, if using internal coordinates, try Cartesian coordinates (
opt=Cartesian) or vice versa. - Advanced Hessian Treatment: In rare cases where the Hessian changes significantly during the optimization, it may be necessary to recalculate it at every step (
opt=calcall). This is computationally expensive but can resolve difficult cases [69]. - Handling Saddle Points: If the optimization converges to a transition state (saddle point with one imaginary frequency), enable automatic restarts. This requires disabling symmetry (
UseSymmetry False), enabling PES point characterization (Properties PESPointCharacter True), and settingMaxRestartsto a value >0 (e.g., 5). The optimizer will then displace the geometry along the imaginary mode and restart [68].
The Scientist's Toolkit: Essential Research Reagents and Parameters
This section details the key "computational reagents" – the parameters and settings that are crucial for a successful geometry optimization experiment.
Table 2: Key parameters and software components for geometry optimization.
| Item/Reagent | Type | Function & Purpose |
|---|---|---|
| Convergence Criteria (Energy, Gradients, Step) | Software Parameter | Defines the termination conditions for the optimization. Tighter criteria yield more precise geometries at the cost of increased computation [68]. |
| Initial Hessian Matrix | Software Parameter/Method | A matrix of second energy derivatives that describes the curvature of the PES. A good initial guess is critical for convergence rate and stability [69]. |
| Coordinate System (Internal, Cartesian) | Software Parameter | The coordinate representation in which the optimization is performed. Internal coordinates are often more efficient for molecules [69]. |
| Berny Optimization Algorithm | Algorithm | The default optimizer in many packages (e.g., Gaussian). It uses and updates the Hessian to efficiently step toward a minimum [69]. |
| PES Point Characterization | Analysis Method | A frequency calculation performed on the optimized structure to confirm it is a minimum (all real frequencies) and not a saddle point [68]. |
| Electronic Structure Method (e.g., DFT, HF) | Quantum Chemical Method | The underlying theory used to calculate the energy and forces for a given nuclear configuration. The choice affects the accuracy and cost of the entire process. |
| Basis Set (e.g., 6-31G*, cc-pVDZ) | Mathematical Basis | A set of functions used to represent molecular orbitals. Larger basis sets provide greater accuracy but increase computational demand significantly. |
Performance Validation: Benchmarking Convergence Efficiency Across Molecular Systems and Methods
The effective management of Distributed Energy Resources (DERs) and the optimization of grid operations are crucial for Distribution System Operators (DSOs) in the context of modern energy systems. The integration of renewable energy sources and electric vehicles (EVs) introduces significant complexity and unpredictability, demanding advanced optimization models that ensure grid stability while minimizing operational costs [4] [54]. Establishing robust validation metrics—convergence speed, stability, and computational cost—is therefore fundamental for evaluating the performance of optimization algorithms and machine learning (ML) models used in this domain. These metrics provide critical insights for benchmarking and guide the development of more efficient and reliable systems for energy grid management, including research on Denial-of-Service (DoS) convergence [54]. This document outlines application notes and experimental protocols for researchers and scientists focused on setting energy grid parameters.
Defining the Core Validation Metrics
The following metrics are essential for a comprehensive evaluation of algorithms in energy grid optimization and related computational tasks.
Table 1: Core Performance and Computational Metrics
| Metric Category | Specific Metric | Definition and Application Context |
|---|---|---|
| Convergence Speed | Number of Epochs/Iterations | The number of complete passes through a dataset or optimization cycles required to reach a satisfactory solution [70]. |
| Time to Convergence | The total computational time (e.g., in seconds) until the algorithm's performance plateaus or meets a predefined threshold. | |
| Stability | PSNR (Peak Signal-to-Noise Ratio) | A metric for evaluating the fidelity of a reconstructed signal or image; used in super-resolution challenges with thresholds like 26.90 dB [71]. |
| Voltage Deviation | A grid-specific metric indicating the stability of the power system; improvements (e.g., 43.5%) demonstrate enhanced grid stability [54]. | |
| Loss Function Plateau | The point in training where the model's loss function ceases to decrease significantly, indicating convergence or a stable state [72] [70]. | |
| Computational Cost | Parameters | The number of trainable parameters in a model (e.g., 0.276 million), indicating model complexity [71]. |
| FLOPs (Floating Point Operations) | The number of floating-point operations required for a single inference, measured for a standard input size (e.g., 16.70 G for a 256x256 image) [71]. | |
| Runtime | The average time required to perform a single inference or a complete optimization cycle (e.g., 22.18 ms) [71]. | |
| Operational Cost | In grid management, the total cost of energy procurement and system operations; models aim for significant reductions (e.g., 19.3%) [54]. |
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Research Reagents and Computational Tools
| Item Name | Function/Application |
|---|---|
| DIV2K & LSDIR Datasets | High-resolution image datasets used for training and validating efficient super-resolution models, providing standardized benchmarks for performance (PSNR) and computational metrics [71]. |
| EFDN (Edge-Enhanced Feature Distillation Network) | A baseline deep learning model that combines re-parameterization and architecture search to achieve a trade-off between performance and computational efficiency [71]. |
| Hiking Optimization Algorithm (HOA) | A metaheuristic optimization algorithm that uses an adaptive search mechanism to explore solution spaces and avoid local optima, effective for multi-objective grid optimization problems [54]. |
| Matbench Discovery | An evaluation framework for benchmarking machine learning models on materials stability predictions, emphasizing prospective validation and task-relevant metrics [73]. |
| Competitive Swarm Optimizer (CSO) & Variants | Evolutionary algorithms designed for solving large-scale multi-objective optimization problems (LSMOPs) through competitive particle update mechanisms [74]. |
| Physics-Informed Neural Networks (PINNs) | Neural networks that incorporate physical laws (e.g., PDEs) into the loss function to solve forward and inverse problems in scientific computing, with convergence speed being a key research focus [72]. |
Experimental Protocols for Metric Validation
Protocol 1: Benchmarking Algorithmic Efficiency
Objective: To quantitatively compare the performance of a novel optimization algorithm against established baselines using convergence, stability, and cost metrics.
Materials: Standard benchmark suites (e.g., LSMOP for optimization, DIV2K for super-resolution), computing cluster with GPU nodes, profiling tools (e.g., PyTorch Profiler).
Methodology:
- Baseline Establishment: Select state-of-the-art algorithms for comparison (e.g., EFDN for super-resolution [71], PSO and KMA for grid optimization [54]).
- Experimental Setup: Implement all algorithms and ensure they are evaluated on the same benchmark datasets or grid simulation environments (e.g., a 33-bus distribution grid model [54]).
- Metric Tracking:
- Convergence Speed: For each run, record the loss function value or primary objective metric at every epoch/iteration. Plot learning curves.
- Stability: For ML models, record validation PSNR at each epoch [71]. For grid optimization, measure final metrics like voltage deviation [54].
- Computational Cost: Use profiling tools to measure the average runtime per iteration and total time to convergence. Calculate FLOPs and parameter count for neural networks [71].
- Statistical Analysis: Execute multiple independent runs with different random seeds. Report the mean and standard deviation for all metrics. Perform statistical significance tests (e.g., t-test) to validate performance differences.
Protocol 2: Training and Validating an Efficient Deep Learning Model
Objective: To train a deep learning model (e.g., for image super-resolution) that meets specific performance thresholds while minimizing computational overhead.
Materials: Training dataset (e.g., DIV2K and LSDIR training splits [71]), validation dataset (e.g., DIV2KLSDIRvalid), deep learning framework (e.g., PyTorch), NVIDIA RTX A6000 GPU or equivalent.
Methodology:
- Model Design: Develop a model architecture aimed at reducing parameters, FLOPs, or runtime relative to a baseline like EFDN [71].
- Training Loop:
- Use an appropriate optimizer (e.g., Adam) and a dynamic learning rate scheduler.
- Perform a forward pass, calculate the loss (e.g., L1 or MSE loss), and update weights via backpropagation.
- Epoch Management: Treat one epoch as a full pass through the training dataset. Implement early stopping with a "patience" parameter (e.g., 10 epochs) to halt training if validation performance does not improve, thus preventing overfitting and saving resources [70].
- Validation and Checkpointing: After each epoch, run inference on the validation set to compute the PSNR. Save the model as a checkpoint only if the validation PSNR improves.
- Final Evaluation: Upon early stopping, load the best checkpoint and run it on the held-out test set to obtain final performance (PSNR) and efficiency (runtime, FLOPs) metrics, ensuring they meet the required thresholds (e.g., >26.99 dB PSNR) [71].
Protocol 3: Multi-Objective Optimization for Grid Parameter Tuning
Objective: To utilize a multi-objective optimization algorithm for setting energy grid parameters that minimize cost and maximize stability, particularly under constraints like EV integration.
Materials: Grid simulation software, a defined multi-objective optimization model (e.g., minimizing energy losses, costs, voltage deviations [54]), Hiking Optimization Algorithm (HOA) implementation [54].
Methodology:
- Problem Formulation: Define the objective function as a weighted sum of targets:
Minimize [Energy Losses, Procurement Costs, Load Shedding, Voltage Deviations, EV/Battery Management Costs]over a 24-hour horizon [54]. - Algorithm Execution: Initialize the HOA with a population of candidate solutions (parameter sets). Iterate until convergence:
- Competitive Search: Use HOA's adaptive mechanism to explore the parameter space, effectively avoiding local optima [54].
- Evaluation: For each candidate solution, run a grid simulation to compute all objective values.
- Solution Selection: After convergence, analyze the Pareto front of non-dominated solutions. Select a final parameter set that offers the best trade-off for the specific grid scenario (e.g., prioritizing voltage stability for DOS convergence research).
- Validation: Compare the performance of the selected solution against a baseline scenario without optimization, reporting percentage improvements in key metrics like operational cost (-19.3%) and voltage deviations (-43.5%) [54].
Workflow and Relationship Visualizations
Experimental Workflow for Model Validation
Stability Assessment Framework
Comparative Analysis of Convergence Performance Across Protein-Ligand Complexes
Accurately predicting the binding affinity between a protein and a small molecule ligand is a fundamental challenge in computational chemistry and drug discovery. The "convergence" of these predictions refers to the stability and reliability of computed free energy values with increased sampling or system refinement. Achieving rapid and robust convergence is critical for practical applications in virtual screening and lead optimization. This analysis examines the convergence performance of various computational methods, focusing on their quantitative accuracy, computational efficiency, and applicability within drug development workflows. Key methodologies include quantum mechanical fragmentation, machine learning-corrected approaches, and hybrid quantum mechanics/molecular mechanics (QM/MM) protocols, each offering distinct trade-offs between computational cost and predictive precision for protein-ligand complexes.
Performance Comparison of Computational Methods
The following table summarizes the convergence performance and key characteristics of various computational methods used for protein-ligand binding affinity prediction and structure determination.
Table 1: Convergence Performance and Characteristics of Protein-Ligand Computational Methods
| Method Name | Reported Performance (R²/MAE/SR) | Computational Cost & Speed | Key Convergence Insight |
|---|---|---|---|
| D3-ML [75] | R² = 0.87 with experiment [75] | Sub-second per complex [75] | Exceptional speed/accuracy balance; dispersion energy central for ranking [75] |
| GMBE-DM [75] | R² = 0.84 with experiment [75] | <5 minutes per complex [75] | Quantum-accurate; systematic improvability; efficient without massive parallelization [75] |
| QM/MM on Multi-Conformers (Qcharge-MC-FEPr) [76] | Pearson R = 0.81; MAE = 0.60 kcal mol⁻¹ [76] | Significantly lower than FEP [76] | High accuracy across diverse targets; uses multi-conformer ensemble for robust convergence [76] |
| Full-Protein QM-PBSA (PBE-D3) [77] | Convergence similar to MM-PBSA at ~50 snapshots [77] | High (2600-atom DFT calculations); viable with HPC [77] | Full-protein DFT energies are highly reproducible; entropy correction requires sufficient sampling (>25 snapshots) [77] |
| Screened Many-Body Expansion (HF-3c) [78] | Reproduces supersystem interaction energies within ~1 kcal/mol [78] | ~1% of conventional supramolecular calculation cost [78] | Two-body calculations with single-residue fragments sufficient for convergent interaction energies [78] |
| Umol (Blind) [79] | Success Rate (SR) = 18% (Ligand RMSD ≤ 2Å) [79] | Not specified | AI-based co-folding from sequence; performance improves with pocket information (SR=45%) [79] |
| AutoDock Vina [79] | Success Rate (SR) = 52% (Ligand RMSD ≤ 2Å) [79] | Fast (classical docking) | High performance dependent on known holo-protein structure; limited flexibility treatment [79] |
| Sfcnn (Deep Learning) [75] | R² = 0.57 with experiment [75] | Fast (ML inference) | Lower transferability across diverse datasets; potential overfitting issues [75] |
Abbreviations: R²: Coefficient of determination; MAE: Mean Absolute Error; SR: Success Rate; RMSD: Root-Mean-Square Deviation.
Experimental Protocols for Key Methods
Protocol 1: Generalized Many-Body Expansion for Density Matrices (GMBE-DM)
Objective: To achieve rapid, quantum-chemically accurate ranking of protein-ligand binding affinities using a density-matrix-based fragmentation approach [75].
Workflow:
- System Preparation: Obtain the 3D structure of the protein-ligand complex. Define the fragmentation scheme, typically partitioning the protein into individual amino acid residues or small peptide segments [75].
- Fragment Generation: Apply the generalized many-body expansion (GMBE) to generate a set of overlapping fragments that collectively represent the entire supersystem.
- Density Matrix Calculation: Perform independent quantum chemical calculations (at a chosen level of theory, such as DFT) on each fragment to compute its density matrix.
- Density Matrix Assembly: Combine the fragment density matrices using set-theoretical principles to construct the full density matrix for the entire protein-ligand complex [75].
- Energy Evaluation: Calculate the total interaction energy from the assembled density matrix. A purification scheme may be applied to ensure N-representability [75].
- Binding Affinity Ranking: Rank the ligands based on the computed interaction energies (ΔEint). The protocol can be run with and without crystal water molecules, and ligand desolvation energy (ΔE(desolv)ligand) can be added for refinement [75].
Protocol 2: QM/MM with Multi-Conformer Free Energy Processing (Qcharge-MC-FEPr)
Objective: To achieve accurate binding free energy estimation by combining QM-derived charges with conformational ensembles from a classical method [76].
Workflow:
- Initial Conformer Sampling: Run the classical "Mining Minima" (MM-VM2) method to generate an ensemble of low-energy conformers and poses for the protein-ligand complex [76].
- Conformer Selection: Select multiple conformers (e.g., up to four) that collectively account for a high percentage (e.g., >80%) of the Boltzmann-weighted probability from the MM-VM2 results [76].
- QM/MM Charge Calculation: For each selected conformer, perform a QM/MM calculation. The ligand is treated with quantum mechanics (QM), while the protein and environment are treated with molecular mechanics (MM). Derive electrostatic potential (ESP) atomic charges for the ligand in the context of the polarized protein environment [76].
- Charge Substitution: Replace the original force field atomic charges of the ligand in each selected conformer with the new QM/MM-derived ESP charges.
- Free Energy Processing (FEPr): Perform the final free energy calculation using the mining minima framework, but using the re-charged conformers. The output is the calculated binding free energy (ΔG_calc) [76].
- Scaling and Validation: Apply a universal scaling factor (USF) of 0.2 to the calculated free energies to offset systematic overestimation and compare the scaled values (ΔGoffset,scaledcalc) with experimental data [76].
Protocol 3: Full-Protein DFT with QM-PBSA
Objective: To compute protein-ligand binding free energies using full-protein Density Functional Theory (DFT) within a QM-PBSA framework [77].
Workflow:
- Molecular Dynamics Sampling: Run a classical molecular dynamics (MD) simulation of the protein-ligand complex in explicit solvent. Use a one-trajectory approach for computational efficiency [77].
- Snapshot Extraction: Extract a series of snapshots (e.g., 50-100) from the equilibrated part of the MD trajectory at regular intervals to represent the conformational ensemble of the bound state [77].
- Full-Protein DFT Single-Point Energy Calculation: For each MD snapshot, perform a full-protein, single-point energy calculation using linear-scaling DFT. This replaces the molecular mechanics gas-phase energy evaluation in traditional MM-PBSA [77].
- Implicit Solvation Calculation: For the same set of snapshots, calculate the polar and non-polar solvation free energies (Gpol and Gnon-pol) using an implicit solvent model (e.g., Poisson-Boltzmann or Generalized Born).
- Entropy Estimation: Calculate the conformational entropy change (TΔS) upon binding, for example, by normal mode or quasi-harmonic analysis. This step is computationally demanding and may require sampling over many snapshots to converge [77].
- Free Energy Averaging: For each snapshot i, compute the free energy Gi = EDFT,i + Gsolvation,i - TΔSi. The final binding free energy is the average over all snapshots: ΔGbind = ⟨GAB⟩ - ⟨GA⟩ - ⟨GB⟩, where the terms for the unbound protein (A) and ligand (B) are obtained by deleting the other component from the complex trajectory [77].
The following diagram illustrates the logical workflow and decision process for selecting an appropriate method based on research goals and constraints.
Diagram 1: A decision workflow for selecting protein-ligand computational methods based on research constraints and objectives.
The Scientist's Toolkit: Essential Research Reagents & Solutions
Table 2: Key Software and Computational Tools for Protein-Ligand Studies
| Tool/Solution Name | Type | Primary Function in Research |
|---|---|---|
| AutoDock-GPU [79] [80] | Docking Software | Generates conformational decoy sets and initial binding poses for ligands within a defined protein binding pocket [80]. |
| Linear-Scaling DFT Code [77] | Quantum Chemistry Software | Enables full-protein Density Functional Theory (DFT) calculations by overcoming traditional cubic scaling, making QM-PBSA feasible [77]. |
| VeraChem VM2 [76] | Free Energy Calculator | Implements the "Mining Minima" (M2) method for classical binding free energy estimation and conformational sampling [76]. |
| RDKit [79] [80] | Cheminformatics Toolkit | Handles ligand preprocessing, SMILES parsing, and molecular validity checks for machine learning and docking pipelines [79] [80]. |
| Umol [79] | AI-Based Prediction | Predicts the fully flexible all-atom structure of a protein-ligand complex directly from protein sequence and ligand SMILES string [79]. |
| Fragment Management Platform [78] | Fragmentation Utility | Manages fragment-based quantum calculations using a screened many-body expansion for convergent interaction energies [78]. |
| PDBbind Database [80] | Curated Dataset | Provides a comprehensive collection of protein-ligand complexes with experimental binding affinity data for method training and validation [80]. |
Benchmarking Different Exchange-Correlation Functionals for Drug-like Molecules
The accuracy of Density Functional Theory (DFT) calculations in simulating drug-like molecules and their interactions with biological targets is critically dependent on the choice of the exchange-correlation (XC) functional [81]. These functionals approximate the quantum mechanical exchange and correlation effects, with different approximations offering varying balances of accuracy and computational cost. For research focused on setting energy grid parameters for density of states (DOS) convergence, selecting an appropriate XC functional is a foundational step, as it directly influences the electronic structure properties obtained from the calculation. This application note provides a structured benchmarking approach and detailed protocols for evaluating XC functionals in the context of drug discovery applications, enabling researchers to make informed decisions tailored to their specific systems and properties of interest.
The development of XC functionals follows a systematic increase in complexity and incorporation of physical ingredients, often referred to as "Jacob's Ladder" [81]. This progression aims to improve accuracy while balancing computational demands.
Table 1: Categories of Exchange-Correlation Functionals
| Functional Type | Key Input Variables | Strengths | Weaknesses | Example Functionals |
|---|---|---|---|---|
| Local Density Approximation (LDA) | Electron density ((n)) | Computationally efficient; foundation for advanced functionals | Systematic over-binding; inaccurate for molecules | SVWN5 [82] [81] |
| Generalized Gradient Approximation (GGA) | (n), Density gradient ((\nabla n)) | Improved molecular geometries and energies | Underestimates band gaps and reaction barriers | PBE [81], BLYP [82] |
| meta-GGA | (n), (\nabla n), Kinetic energy density ((\tau)) | Better for diverse properties (e.g., solid-state) | Higher computational cost than GGA | SCAN [81], B97M-V [82] |
| Hybrid | Mix of HF and semilocal exchange | Improved accuracy for molecular energies & properties | High computational cost (HF exchange) | B3LYP [83], PBE0 [82] |
| Range-Separated Hybrid | HF/semilocal exchange split by range | Improved electronic properties; faster convergence in solids | Parameter choice (ω) can be system-specific | HSE06 [83], ωB97M-D3(BJ) [84] |
Figure 1: The "Jacob's Ladder" hierarchy of DFT functionals, illustrating increasing complexity and physical ingredients.
Critical Datasets for Benchmarking Drug-Like Molecules
The creation of high-quality, specialized datasets has been pivotal for the rigorous development and testing of XC functionals for biochemical systems. These datasets provide reference quantum chemistry calculations for training and validation.
The SPICE Dataset
The SPICE (Small-molecule/Protein Interaction Chemical Energies) dataset is a quantum chemistry dataset specifically designed for training and testing potentials relevant to simulating drug-like small molecules interacting with proteins [84]. Its design fulfills several critical requirements for meaningful benchmarking in this domain:
- Broad Chemical Space: It covers 15 elements, including both charged and uncharged molecules, sampling a wide range of covalent and non-covalent interactions found in drug-like molecules and proteins [84].
- Relevant Conformational Sampling: It includes over 1.1 million molecular conformations, extending beyond minimum-energy structures to cover the range of conformations encountered in molecular dynamics simulations and conformational searches [84].
- High-Quality Reference Data: Energies and forces are calculated at the ωB97M-D3(BJ)/def2-TZVPPD level of theory, a high-level meta-GGA functional with dispersion correction, providing a robust benchmark [84].
- Rich Data Outputs: The dataset provides not only energies and forces but also other useful quantities like multipole moments and bond orders, enabling multi-faceted benchmarking [84].
Other Relevant Datasets
Other datasets exist but have limitations for drug-discovery applications. OrbNet Denali is large and chemically diverse but provides only energies, not forces, limiting its information content [84]. QMugs contains diverse molecules but only in their energy-minimized conformations, making it unsuitable for dynamics [84]. The ANI series (ANI-1, ANI-1x, ANI-1ccx) is extremely large but covers only four elements and no charged molecules, which is insufficient for modeling proteins or many drug molecules [84].
Quantitative Benchmarking of Popular XC Functionals
The performance of an XC functional can vary significantly depending on the target property. Benchmarking against high-quality reference data is therefore essential.
Table 2: Functional Performance for Key Properties
| Functional | Type | Band Gap Accuracy (Solids) [83] | General Molecular Accuracy | Dispersion Treatment | Computational Cost |
|---|---|---|---|---|---|
| PBE | GGA | Poor (systematic underestimation) | Good geometries; moderate energies | None (requires add-on) | Low |
| B3LYP | Global Hybrid | Moderate | Good for organic molecules | None (requires add-on) | High |
| HSE06 | Range-Separated Hybrid | High | Good for solids & molecules | None (requires add-on) | High (less than B3LYP in solids) |
| mBJ | meta-GGA | Very High | Designed for band gaps | None | Moderate |
| ωB97M-D3(BJ) | Range-Separated Hybrid + Dispersion | N/A (Used for SPICE benchmark) | High (used for SPICE dataset) | Excellent (built-in D3(BJ)) | Very High |
| SCAN | meta-GGA | Moderate to High | Broadly accurate for diverse systems | Moderate (meta-GGA) | Moderate |
Detailed Experimental Protocols for Benchmarking
A robust benchmarking procedure involves multiple stages, from initial system preparation to the final analysis of results against reference data. The following workflow and protocols outline this process.
Figure 2: A high-level workflow for benchmarking exchange-correlation functionals.
Protocol 1: Benchmarking against the SPICE Dataset
This protocol uses the SPICE dataset to evaluate the performance of different XC functionals for calculating energies and forces of drug-like molecules and peptide interactions [84].
5.1.1 Research Reagent Solutions
| Item / Resource | Function in Benchmarking |
|---|---|
| SPICE Dataset | Provides reference energies, forces, and other properties for a diverse set of small molecules, dimers, dipeptides, and solvated amino acids [84]. |
| Quantum Chemistry Software | Software (e.g., Gaussian, Q-Chem, ORCA) is used to compute the properties of the molecules in the dataset using the XC functionals being tested. |
| Machine Learning Potential Framework | Tools (e.g., ANI, SchNet) can be used to train potentials on the SPICE data, allowing for efficient evaluation of functional accuracy across chemical space [84]. |
| ωB97M-D3(BJ)/def2-TZVPPD | Serves as the high-level reference theory against which the performance of other, less expensive functionals is compared [84]. |
5.1.2 Step-by-Step Procedure
- Data Acquisition and Partitioning: Download the latest versioned release of the SPICE dataset from its public repository. Partition the data into training/validation/test sets, ensuring that molecules in the test set are not represented in the training set to avoid biased results.
- Selection of XC Functionals: Choose a diverse set of functionals for testing, representing different rungs of Jacob's Ladder (e.g., PBE (GGA), B97M-V (meta-GGA), B3LYP (hybrid), ωB97M-D (range-separated hybrid)).
- Computational Setup: For each molecule and conformation in the SPICE test set, perform a single-point energy and force calculation using each of the selected XC functionals. Employ a consistent, high-quality basis set (e.g., def2-TZVP) for all calculations.
- Error Metrics Calculation: For each XC functional, compute error metrics relative to the SPICE reference data across the entire test set. Key metrics include:
- Mean Absolute Error (MAE) of energies per atom.
- Root Mean Square Error (RMSE) of forces.
- Relative energy errors for conformational rankings.
- Performance Analysis: Rank the tested XC functionals based on the calculated error metrics. Identify which functional provides the best trade-off between accuracy and computational cost for the target properties (e.g., energy, forces, or both).
Protocol 2: Assessing Electronic Properties for DOS Convergence
This protocol is specifically designed for the context of setting energy grid parameters for DOS convergence research. It benchmarks XC functionals based on their ability to reproduce accurate electronic structures.
5.2.1 Research Reagent Solutions
| Item / Resource | Function in Benchmarking |
|---|---|
| Solid-State Band Gap Datasets | Curated sets of materials with experimentally measured band gaps provide a benchmark for evaluating a functional's ability to reproduce electronic DOS and band structures [83]. |
| Projected Density of States (PDOS) | A computational output that projects the DOS onto atomic orbitals, crucial for understanding electronic contributions in complex systems like organometallic drugs. |
| Tuned Range-Separation Parameter (ω) | In range-separated hybrids, a system-specific ω parameter can be optimized non-empirically to satisfy DFT conditions, improving the accuracy of frontier orbital energies [85]. |
5.2.2 Step-by-Step Procedure
- System Selection: Construct a test set of molecules and materials relevant to your research. This should include typical fragments from your drug-like molecules and, if applicable, any periodic structures.
- Reference Data Collection: For the test set, gather reliable experimental or high-level theoretical data for electronic properties. This can include fundamental band gaps from experiments [83] or ionization potentials/electron affinities from wavefunction methods.
- Electronic Structure Calculation: For each system in the test set, perform a geometry optimization followed by a single-point calculation with a dense k-point mesh (for solids) or a large basis set (for molecules) using each XC functional under test.
- DOS and Band Gap Extraction: Calculate the total and projected DOS for each system. Extract the Kohn-Sham band gap (or HOMO-LUMO gap for molecules) from the DOS output.
- Accuracy Assessment and Functional Selection:
- Calculate the MAE and RMSE of the calculated band gaps against the reference data.
- Analyze the shape and character of the PDOS for known systems to check for qualitative correctness (e.g., correct orbital ordering).
- Based on the accuracy in reproducing gaps and PDOS features, select the functional that is most reliable for subsequent DOS convergence tests.
Benchmarking exchange-correlation functionals is a critical step in establishing a reliable computational framework for studying drug-like molecules, particularly for specialized goals like achieving DOS convergence. The hierarchical nature of functionals means there is no universal "best" choice; the optimal functional depends on the specific property of interest and the available computational resources. Leveraging modern, chemically diverse datasets like SPICE allows for a comprehensive evaluation of functional performance across a region of chemical space directly relevant to drug discovery. By following the structured protocols outlined here, researchers can make justified decisions on XC functionals, thereby ensuring the robustness and accuracy of their subsequent research on energy grid parameters and electronic properties.
Validation Protocols for Ensuring Result Reliability Post-Convergence
In energy grid parameter research, achieving algorithmic convergence for Distribution System Operator (DSO) models represents merely the initial step toward obtaining actionable insights. Post-convergence validation encompasses the systematic methodologies and protocols employed to verify that optimized results are not merely mathematical artifacts but reliable, robust, and physically plausible solutions to the underlying grid management problem. The transition toward complex, multi-objective optimization in modern grid systems—integrating distributed energy resources (DERs), electric vehicles (EVs), and variable renewable generation—has rendered rigorous validation protocols indispensable for ensuring operational reliability and informing critical infrastructure decisions [4] [54].
The core challenge addressed by these protocols is the inherent trade-off between model fidelity and computational tractability. Sophisticated grid optimization algorithms, including metaheuristics like the Hiking Optimization Algorithm (HOA) or meta-model-based approaches, may converge to solutions that are optimal within the constrained mathematical framework but potentially vulnerable to real-world uncertainties, data inaccuracies, or unmodeled physical constraints [86] [54]. Consequently, a robust validation framework must assess result stability, sensitivity to input parameters, and consistency with established grid physics to ensure that proposed parameter sets for DSO convergence will translate into safe, efficient, and resilient grid operations.
Core Principles and Quantitative Benchmarks for Validation
Effective validation is anchored upon three foundational pillars: Stability, Sensitivity, and Plausibility. Stability verification ensures that the converged solution is not a fragile local optimum and that re-initialization or minor perturbations do not lead to drastically different outcomes. Sensitivity analysis quantifies how variations in input parameters (e.g., load forecasts, renewable generation profiles, market prices) propagate through the model to affect the final results, identifying critical parameters that demand high accuracy. Plausibility checking guarantees that the optimized parameters adhere to the physical and operational laws of the grid, such as power flow constraints and voltage limits [86].
Validation requires benchmarking against quantitative metrics to objectively assess the reliability of converged results. The following table summarizes the key metrics and their target benchmarks derived from current literature on grid optimization.
Table 1: Key Quantitative Metrics for Post-Convergence Validation
| Metric Category | Specific Metric | Validation Benchmark & Target | Interpretation |
|---|---|---|---|
| Statistical Reliability | Coefficient of Variation (CV) of Objective Function | CV < 2% over multiple runs [86] | Indicates high solution stability. |
| Confidence Interval for Key Outputs | 95% CI within ±0.5% of mean value [86] | Quantifies uncertainty in results. | |
| Performance Verification | Voltage Deviation | Minimization; e.g., >43.5% improvement from baseline [54] | Confirms grid stability and power quality. |
| Energy Losses | Minimization; e.g., >59.7% reduction from baseline [54] | Validates economic and technical efficiency. | |
| Model Fidelity | Meta-model R² (Goodness-of-Fit) | R² > 0.95 against full simulation model [86] | Ensures surrogate model accuracy. |
| Mean Absolute Percentage Error (MAPE) | MAPE < 5% for key dependent variables [86] | Validates forecasting/prediction accuracy. |
Detailed Experimental Validation Protocols
Protocol 1: Stochastic Robustness and Multi-Start Analysis
1. Objective: To verify that the converged solution is a stable, global optimum rather than a local optimum, and to assess its reliability under different algorithmic initializations.
2. Methodology:
- Execute a minimum of 50 independent optimization runs from stochastically generated initial points within the feasible parameter space.
- For each run, record the final converged value of the primary objective function (e.g., total operational cost) and the resulting DSO parameter set.
- Compute the mean, standard deviation, and coefficient of variation (CV) of the objective function values across all runs.
- Perform a cluster analysis on the resulting parameter sets to identify if runs converge to a single, tight cluster or multiple, disparate clusters.
3. Data Analysis & Interpretation:
- A low CV (<2%) and a single dominant cluster of parameters indicate a robust, reliable optimum [86].
- A high CV or multiple clusters suggest the presence of numerous local optima, casting doubt on the reliability of any single result and necessitating the use of global optimization techniques with enhanced exploration capabilities.
Protocol 2: Uncertainty and Sensitivity Analysis (UA/SA)
1. Objective: To identify which input parameters most significantly influence the optimized outcome, thereby guiding data acquisition efforts and highlighting potential operational risks.
2. Methodology:
- Define a plausible range of variation (e.g., ±10%) for all critical input parameters, including load forecasts, renewable generation (PV/wind) forecasts, energy pricing data, and EV charging demand [54].
- Employ a quasi-Monte Carlo sampling method or a structured design-of-experiments (DoE) approach to efficiently explore the high-dimensional input space [86].
- For each sample set of inputs, run the simulation model with the previously optimized DSO parameters fixed and record the outputs.
- Calculate global sensitivity indices (e.g., Sobol indices) to quantify the fraction of output variance attributable to each input parameter and their interactions.
3. Data Analysis & Interpretation:
- Parameters with high sensitivity indices are prioritized for accurate forecasting and real-time monitoring.
- This analysis reveals the "risk profile" of the optimized solution, showing how robust the performance is expected to be in the face of inherent forecast uncertainties.
Table 2: Key Input Parameters for Sensitivity Analysis in EV-Integrated Grids
| Input Parameter | Typical Range for SA | Primary Outputs Affected | Mitigation Strategy if Highly Sensitive |
|---|---|---|---|
| EV Charging Demand | ±15% of forecast [54] | Energy losses, voltage deviation, load shedding | Implement smart charging with real-time adjustment. |
| Solar PV Generation | ±20% of forecast [54] | Energy procurement cost, PV curtailment | Deploy complementary fast-ramping storage. |
| Real-Time Electricity Price | ±10% of forecast [54] | Total operational cost | Utilize financial hedging instruments. |
| Distribution Load | ±5% of forecast [86] | Voltage profile, energy losses | Enhance load forecasting with machine learning. |
Protocol 3: Cross-Model Verification and Physical Plausibility Check
1. Objective: To ensure that results are not an artifact of a specific modeling choice and that they adhere to the physical laws governing the power grid.
2. Methodology:
- Cross-Model Verification: Validate the optimized parameters by running them in an independent, high-fidelity simulation environment (e.g., a full-order model in EnergyPlus or a detailed power flow solver) that was not used during the optimization process. Compare key outputs like power flows and voltage levels [86].
- Dynamic Simulation: Test the proposed parameters under a set of representative dynamic grid events (e.g., a sudden drop in solar generation, a distribution line fault, a rapid surge in EV charging) to ensure the system remains stable and within safe operating limits.
- Constraint Audit: Perform a comprehensive review to ensure no soft constraints were critically violated and that all hard constraints (e.g., thermal limits of cables, voltage bounds) are fully respected.
3. Data Analysis & Interpretation:
- Significant discrepancies between the optimization model's predictions and the high-fidelity model's outputs indicate a potential flaw in the optimization problem formulation or the underlying meta-model.
- Successful passage of dynamic and constraint tests provides high confidence that the parameters are physically viable and safe for implementation.
Diagram 1: Core validation workflow post-convergence, illustrating the sequential protocol for ensuring result reliability.
The Scientist's Toolkit: Essential Research Reagents and Solutions
The experimental protocols outlined above rely on a suite of computational tools and datasets, which function as the essential "research reagents" in the domain of energy grid parameter research.
Table 3: Key Research Reagent Solutions for Grid Parameter Validation
| Reagent / Tool | Function / Purpose | Exemplars & Notes |
|---|---|---|
| High-Fidelity Grid Simulator | Serves as the ground-truth benchmark for validating results from optimization models. Provides detailed physical representation. | EnergyPlus, OpenDSS, GridLAB-D, MATPOWER. Critical for Cross-Model Verification. |
| Uncertainty & Sensitivity Analysis Library | Automates the computation of sensitivity indices and manages quasi-Monte Carlo sampling. | Integrated Global Sensitivity and Uncertainty Management software [86], SALib (Python). |
| Meta-Modeling Framework | Creates fast, analytic approximations of complex simulation models to enable efficient optimization and vast parameter exploration. | Polynomial Chaos Expansion, Gaussian Process Regression, Neural Networks. Requires R² > 0.95 [86]. |
| Benchmark Grid Datasets | Provides standardized, realistic network and load data for reproducible testing and validation of optimization algorithms. | IEEE 33-bus, 123-bus test feeders; IoT-Enabled Smart Grid Dataset [87]. |
| Performance Profiling Toolkit | Measures computational efficiency (e.g., time per simulation, memory footprint) alongside solution quality. | Custom scripts to track convergence time, number of function evaluations, and parallel scaling. |
Advanced Topics: Security and Resilience Validation
With the increasing interconnectivity of smart grids, validating the resilience of optimized parameters against cyber threats has become a critical extension of traditional protocols. This involves stress-testing DSO parameters and control logic under various cyber-attack scenarios, such as False Data Injection (FDI) into sensor readings or Distributed Denial of Service (DDoS) attacks on communication networks [88] [87].
Validation protocols here incorporate anomaly detection models, often based on machine learning or federated learning frameworks, to identify deviations from expected operation caused by such attacks. The reliability of a result is therefore not only its static optimality but also its inherent robustness within a contested cyber-physical environment. This requires simulating adversarial actions and verifying that the system's response, guided by the optimized parameters, either remains stable or fails gracefully without catastrophic consequences [88].
Diagram 2: The feedback loop from sensitivity analysis to resilient system design, turning validation insights into actionable design improvements.
Application Note: Convergence of QSP and Pharmacometrics in Oncology Drug Development
This application note details the successful implementation of convergence research methodologies to address the high failure rates in oncology drug development. By integrating Quantitative Systems Pharmacology (QSP) with traditional pharmacometrics, researchers have achieved more predictive modeling of drug efficacy and safety profiles, particularly for molecular targets with complex biological context. The convergence approach has demonstrated potential to reduce late-stage attrition through improved trial design and patient stratification strategies [89].
Table 1: Performance Metrics of Convergence Approaches in Pharmaceutical Development
| Convergence Approach | Traditional Success Rate | Convergence Success Rate | Development Time Impact | Key Performance Indicators |
|---|---|---|---|---|
| QSP-Pharmacometrics Integration | 6.7% (Phase 1) | 14.2% (Projected) | Reduction of 6-9 months | 35% improvement in PK/PD prediction accuracy [89] |
| AI-Nanoparticle Diagnostics | N/A | 89% biomarker detection accuracy | Real-time profiling | 72-hour diagnostic turnaround versus 3-4 weeks conventional [90] |
| Master Protocol Implementation | 10% (Historical average) | 16.8% (Adaptive trials) | 40% patient enrollment acceleration | 30% cost reduction per evaluable patient [91] |
Table 2: Economic Impact of Convergence Strategies in Pharmaceutical R&D
| Strategy | Pre-Convergence R&D ROI | Post-Implementation ROI | Regulatory Submission Efficiency | Patent Quality Improvement |
|---|---|---|---|---|
| Data Intelligence Convergence | 4.1% | 8.7% (Projected) | 25% faster agency review cycles | 45% increase in patent citations [92] |
| Cross-Disciplinary Platform Development | $350B patent cliff exposure | 22% risk mitigation | 18-month earlier lifecycle planning | 30% broader patent fortress coverage [92] |
Experimental Protocols
Protocol: Integrated QSP-Pharmacometrics Workflow for Dose Optimization
Purpose and Scope
This protocol describes a sequential integration methodology for combining QSP and pharmacometrics to optimize dosing regimens for novel oncology therapeutics, specifically designed for molecular targets with narrow therapeutic windows [89].
Materials and Equipment
- Computational Environment: End-to-end computation platform with parallel processing capability
- Software Tools: MATLAB, R, NONMEM, or equivalent pharmacometric software
- Data Infrastructure: Secure data repository for clinical, genomic, and biomarker data
- Validation Framework: Machine learning algorithms for model cross-validation
Procedure
QSP Model Initialization (Days 1-30)
- Develop mechanistic physiology model incorporating disease pathophysiology
- Define system parameters using in vitro and preclinical data
- Establish baseline system behavior in healthy and disease states
Pharmacometric Interface (Days 31-60)
- Implement structural PK/PD model using population approaches
- Identify covariates influencing drug exposure and response
- Characterize between-subject and within-subject variability
Cross-Informative Validation (Days 61-75)
- Utilize QSP simulations to inform pharmacometric priors
- Apply pharmacometric population estimates to refine QSystem parameters
- Conduct virtual population trials to assess trial design options
Parallel Synchronization (Days 76-90)
- Execute simultaneous QSP and pharmacometric analyses
- Compare outcomes across multiple uncertainty scenarios
- Generate integrated dose recommendation with confidence intervals
Data Analysis
The convergence advantage is quantified through improved precision of target engagement estimates and reduced uncertainty in therapeutic index projections. Success metrics include ≥30% improvement in clinical endpoint prediction accuracy and ≥25% reduction in required sample size for proof-of-concept studies [89].
Protocol: AI-Enhanced Nanoparticle Drug Delivery for Heterogeneous Tumors
Purpose and Scope
This protocol addresses cancer heterogeneity through convergent artificial intelligence and nanotechnology approaches, enabling patient-specific biomarker sensing and targeted drug delivery for complex solid tumors with evolving resistance mechanisms [90].
Materials and Equipment
- Nanoparticle Systems: Gold nanoparticles (AuNPs), polymeric nanoparticles, liposomes
- Characterization Equipment: Dynamic light scattering, electron microscopy
- AI/ML Platform: TensorFlow or PyTorch with biomedical imaging extensions
- In vitro Models: 3D tumor spheroids, organ-on-chip systems
- In vivo Models: Patient-derived xenografts with humanized components
Procedure
Biomarker Identification Phase (Days 1-21)
- Train deep learning algorithms on multi-omics patient data
- Identify predictive biomarkers for drug sensitivity and resistance
- Validate biomarker candidates using in silico perturbation analysis
Nanoparticle Design Optimization (Days 22-50)
- Engineer nanoparticle surfaces for active targeting of identified biomarkers
- Optimize drug loading capacity and release kinetics
- Validate targeting specificity using flow cytometry and imaging
AI-Mediated Delivery Prediction (Days 51-70)
- Input patient-specific imaging and genomic data into predictive algorithm
- Simulate nanoparticle distribution and tumor penetration
- Predict optimal dosing schedule and combination therapies
Therapeutic Efficacy Assessment (Days 71-100)
- Evaluate targeted delivery efficiency in heterogeneous tumor models
- Quantify biomarker modulation and pathway engagement
- Assess therapeutic index improvement over conventional administration
Data Analysis
The convergence efficacy is measured through enhanced localization metrics (≥3.5-fold improvement in tumor-to-normal tissue ratio) and superior therapeutic outcomes (≥40% increase in progression-free survival in preclinical models) compared to non-targeted approaches [90].
Visualization of Convergence Methodologies
QSP-Pharmacometrics Integration Workflow
AI-Nanoparticle Convergence Framework
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Research Reagents and Materials for Convergence Pharmaceutical Research
| Reagent/Material | Function | Application in Convergence Research | Key Suppliers |
|---|---|---|---|
| QSP Modeling Platforms | Mechanistic simulation of disease pathophysiology | Predicts system-level drug effects; integrates with pharmacometric models [89] | Certara, R, MATLAB |
| Population PK/PD Software | Quantifies variability in drug exposure and response | Provides population parameter estimates for QSP model refinement [89] | NONMEM, Monolix |
| Multifunctional Nanoparticles | Targeted drug delivery and biomarker sensing | Enables localized therapy and real-time treatment monitoring [90] | Various specialized manufacturers |
| AI/ML Algorithm Suites | Analysis of complex biomedical datasets | Identifies biomarkers, predicts drug interactions, optimizes nanocarriers [90] | TensorFlow, PyTorch |
| Master Protocol Templates | Framework for adaptive clinical trials | Enables efficient evaluation of multiple therapies within unified structure [91] | FDA guidance-based templates |
| Data Interoperability Tools | Integration of disparate data sources | Enables convergence of patent, clinical, and scientific data streams [92] | Custom informatics platforms |
The case studies presented demonstrate that convergence research approaches yield substantial improvements in addressing challenging pharmaceutical targets. Through strategic integration of computational, technological, and regulatory methodologies, researchers can achieve enhanced predictive capability and development efficiency. The documented protocols provide implementable frameworks for extending these convergence advantages across the drug development pipeline, potentially mitigating the industry's productivity challenges while improving the precision of therapeutic interventions [89] [90] [92].
Conclusion
Achieving reliable SCF convergence is fundamental to accurate quantum chemical calculations in drug discovery, directly impacting the prediction of molecular properties, binding affinities, and reaction mechanisms. This comprehensive analysis demonstrates that successful convergence requires integrated strategies combining appropriate method selection, careful parameter tuning, systematic troubleshooting, and rigorous validation. The interplay between algorithmic choices, numerical settings, and molecular system characteristics necessitates a nuanced approach tailored to specific research contexts. Future directions should focus on developing more robust adaptive convergence algorithms, machine learning-enhanced parameter optimization, and specialized protocols for challenging pharmaceutical compounds like metalloenzyme inhibitors. As computational drug discovery advances toward increasingly complex biological systems, mastering SCF convergence will remain crucial for generating reliable, predictive results that accelerate therapeutic development.
References
- 1. A critical review of machine learning interatomic potentials ... [https://www.oaepublish.com/articles/jmi.2025.17]
- 2. SCF — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/SCF.html]
- 3. Open-Source Projects We Wish Existed - Rowan Scientific [https://rowansci.com/blog/open-source-projects-we-wish-existed]
- 4. Empowering distribution system operators: A review of ... [https://www.sciencedirect.com/science/article/pii/S2405844024108316]
- 5. Enhancing Smart Grid Security and Efficiency: AI, Energy ... [https://www.mdpi.com/1996-1073/18/17/4747]
- 6. Introducing Open Molecules 2025: A massive dataset for ... [https://www.linkedin.com/posts/samuel-blau_the-open-molecules-2025-dataset-is-out-with-activity-7328461647461654529-ToOx]
- 7. EMFF-2025: a general neural network potential for ... [https://www.nature.com/articles/s41524-025-01809-w]
- 8. Comprehensive framework for smart residential demand ... [https://www.nature.com/articles/s41598-025-93817-5]
- 9. An Overview of Self-Consistent Field Calculations Within ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7179435/]
- 10. 4.1 Overview‣ Chapter 4 Self-Consistent Field ... [https://manual.q-chem.com/5.3/Ch4.S1.html]
- 11. Advancing convergence research: Renewable energy ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9897475/]
- 12. Exploring Meta's Open Molecules 2025 (OMol25) & Universal ... [https://www.rowansci.com/blog/exploring-open-molecules-2025]
- 13. Active Learning Exploration of Transition-Metal Complexes ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9976347/]
- 14. Insights from hybrid QM/MM MD simulations [https://www.sciencedirect.com/science/article/abs/pii/S002195172500586X]
- 15. End to end drug discovery: Revolutionizing 2025 [https://lifebit.ai/blog/end-to-end-drug-discovery/]
- 16. The Drug Development Timeline: Why It Takes Over 10 Years [https://intuitionlabs.ai/articles/drug-development-timeline-explained]
- 17. AI-Driven Drug Discovery 2025: How Pharma Giants Are ... [https://www.datamintelligence.com/blogs/ai-driven-drug-discovery-2025-how-pharma-giants-are-cutting-rd-timelines-by-50]
- 18. Unlocking the potential: multimodal AI in biotechnology ... [https://www.nature.com/articles/s41746-025-01992-6]
- 19. How Modern Biotech Is Revolutionizing Drug Discovery in ... [https://federated-learning.sherpa.ai/en/blog/biotechnology-accelerating-drug-discovery]
- 20. Converging global crises are forcing the rapid adoption of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8129828/]
- 21. What experts at Flatiron, PicnicHealth, MMIT expect for 2025 [https://www.drugdiscoverytrends.com/clinical-care-convergence-2025/]
- 22. Leading AI-Driven Drug Discovery Platforms [https://www.sciencedirect.com/science/article/abs/pii/S0031699725075118]
- 23. The Convergence Advantage: A Framework for Integrating ... [https://www.drugpatentwatch.com/blog/the-convergence-advantage-a-framework-for-integrating-patent-clinical-and-scientific-data-to-drive-life-sciences-innovation-strategy/?srsltid=AfmBOopJ_qJKGTYPCmy-4OA0XPfxhxfrtrlBw3frOVP5nR7LAokRR4M2]
- 24. Top 5 Drug Discovery Trends 2025 Driving Breakthroughs [https://www.pelagobio.com/cetsa-drug-discovery-resources/blog/drug-discovery-trends-2025/]
- 25. Density functional theory [https://en.wikipedia.org/wiki/Density_functional_theory]
- 26. Self Consistent Field (SCF) — BAND 2025.1 documentation [https://www.scm.com/doc/BAND/Accuracy_and_Efficiency/SCF.html]
- 27. Geometry Optimization troubleshooting — ADF 2025.1 ... - SCM [https://www.scm.com/doc/ADF/Rec_problems_questions/Geometry_Optimization.html]
- 28. 4.5.2 Basic Convergence Control Options [https://manual.q-chem.com/6.3/Ch4.S5.SS2.html]
- 29. Density Functional (DFT) Methods [https://gaussian.com/dft/]
- 30. Density functional theory (DFT) [https://pyscf.org/user/dft.html]
- 31. DFT [https://www.cup.uni-muenchen.de/ch/compchem/energy/dft1.html]
- 32. SCF Convergence Guidelines for ADF - SCM [https://www.scm.com/doc/ADF/Rec_problems_questions/SCF.html]
- 33. The self-consistent-field cycle - The SIESTA project [https://docs.siesta-project.org/projects/siesta/en/stable/tutorials/basic/scf-convergence/]
- 34. The self-consistent-field cycle - The SIESTA project [https://docs.siesta-project.org/projects/siesta/en/5.0/tutorials/basic/scf-convergence/]
- 35. Increasing k-point grid to take DOS calculation [https://mattermodeling.stackexchange.com/questions/5045/increasing-k-point-grid-to-take-dos-calculation]
- 36. Converging SCF [http://abacus.deepmodeling.com/en/latest/advanced/scf/converge.html]
- 37. Realistic finite temperature simulations for the magnetic and ... [https://pubs.rsc.org/en/content/articlehtml/2025/tc/d5tc01141h]
- 38. How to analyse a density of states [https://www.sciencedirect.com/science/article/pii/S277294942200002X]
- 39. Comparison of Two Convergence Criterion in the ... [https://www.mdpi.com/2073-4441/14/19/2952]
- 40. Can adaptive grid refinement produce grid-independent ... [https://www.sciencedirect.com/science/article/abs/pii/S0021999117303650]
- 41. Grid is Good. Adaptive Refinement Algorithms for Off-the-Grid ... [https://ojmo.centre-mersenne.org/articles/10.5802/ojmo.39/]
- 42. Convergence for the Wang-Landau density of states [https://impact.ornl.gov/en/publications/convergence-for-the-wang-landau-density-of-states/]
- 43. Digitalization technology based research on intelligent ... [https://www.nature.com/articles/s41598-025-99082-w]
- 44. A Temperature Prediction Model for Flexible Electronic ... [https://www.mdpi.com/2072-666X/15/4/430?]
- 45. The Conundrum of Diffuse Basis Sets: A Blessing for ... [https://arxiv.org/html/2502.04631v1]
- 46. The vDZP Basis Set Is Effective For Many Density Functionals [https://www.rowansci.com/publications/vdzp]
- 47. What considerations must be made when selecting a basis ... [https://mattermodeling.stackexchange.com/questions/13987/what-considerations-must-be-made-when-selecting-a-basis-set]
- 48. The QDπ dataset, training data for drug-like molecules and ... [https://www.nature.com/articles/s41597-025-04972-3]
- 49. Ten quick tips to perform meaningful and reproducible ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12064015/]
- 50. A practical guide to large-scale docking [https://www.nature.com/articles/s41596-021-00597-z]
- 51. Pharmacophore-Guided Generative Design of Novel Drug- ... [https://arxiv.org/html/2510.01480v1]
- 52. An integrated approach for oscillation diagnosis in linear ... [https://www.sciencedirect.com/science/article/abs/pii/S0263876214003062]
- 53. Routing protocol design guidelines for smart grid ... [https://www.sciencedirect.com/science/article/abs/pii/S1389128613003927]
- 54. A multi-objective optimization framework for EV-integrated ... [https://www.nature.com/articles/s41598-025-97271-1]
- 55. Troubleshooting — BAND 2025.1 documentation - SCM [https://www.scm.com/doc/BAND/Troubleshooting/Trouble_Shooting.html]
- 56. 7.7. SCF Convergence - ORCA 6.0 Manual [https://www.faccts.de/docs/orca/6.0/manual/contents/detailed/scfconv.html]
- 57. 2.6. Self-Consistent-Field (SCF) [https://orca-manual.mpi-muelheim.mpg.de/contents/essentialelements/scf.html]
- 58. Setting up SCF parameters [https://www.tcm.phy.cam.ac.uk/castep/documentation/WebHelp/content/modules/castep/tskcastepsetelecscf.htm]
- 59. Convergence Questions [https://downloads.wavefun.com/FAQ/converge.html]
- 60. BasisSet — | QuantumATKX-2025.06 Documentation [https://docs.quantumatk.com/manual/Types/BasisSet/BasisSet.html]
- 61. Approaching the basis set limit in Gaussian-orbital-based ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8556001/]
- 62. Numerical Integration — ADF 2025.1 documentation - SCM [https://www.scm.com/doc/ADF/Input/Numerical_integration.html]
- 63. Integral [https://thiele.ruc.dk/~spanget/help/k_integral.htm]
- 64. G09 Keyword: Integral [http://sobereva.com/g09/k_integral.htm]
- 65. The density functional program [https://www.molpro.net/manual/doku.php?id=the_density_functional_program]
- 66. Abstraction hierarchy to define biofoundry workflows and ... [https://www.nature.com/articles/s41467-025-61263-6]
- 67. Multi-level meta-workflows: new concept for regularly ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5073744/]
- 68. Geometry optimization — AMS 2025.1 documentation - SCM [https://www.scm.com/doc/AMS/Tasks/Geometry_Optimization.html]
- 69. Geometry Optimization - Basic Considerations [https://www.cup.uni-muenchen.de/ch/compchem/geom/basic.html]
- 70. Epochs in day-to-day machine learning processes [https://nebius.com/blog/posts/epochs-in-day-to-day-ml-pipelines]
- 71. The Tenth NTIRE 2025 Efficient Super-Resolution ... [https://arxiv.org/html/2504.10686v1]
- 72. Enhancing convergence speed with feature enforcing ... [https://www.nature.com/articles/s41598-024-74711-y]
- 73. A framework to evaluate machine learning crystal stability ... [https://www.nature.com/articles/s42256-025-01055-1]
- 74. A multi-stage competitive swarm optimization algorithm for ... [https://www.sciencedirect.com/science/article/abs/pii/S0957417424022784]
- 75. Accurate and Rapid Ranking of Protein–Ligand Binding ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12503914/]
- 76. Accurate protein-ligand binding free energy estimation ... [https://www.nature.com/articles/s42004-024-01328-7]
- 77. Protein–ligand free energies of binding from full ... [https://pubs.rsc.org/en/content/articlehtml/2021/cp/d1cp00206f]
- 78. Convergent Protocols for Computing Protein-Ligand ... - PubMed [https://pubmed.ncbi.nlm.nih.gov/39745995/]
- 79. Structure prediction of protein-ligand complexes from ... [https://www.nature.com/articles/s41467-024-48837-6]
- 80. Accurate prediction of protein–ligand interactions by ... [https://jcheminf.biomedcentral.com/articles/10.1186/s13321-024-00912-2]
- 81. Category: Exchange - correlation - VASP Wiki functionals [https://www.vasp.at/wiki/XC_functionals?title=XC_functionals]
- 82. 5.3.4 Exchange - Correlation ‣ 5.3 Overview of Available... Functionals [https://manual.q-chem.com/5.2/Ch5.S3.SS4.html]
- 83. Exchange-correlation functionals for band gaps of solids [https://www.nature.com/articles/s41524-020-00360-0]
- 84. SPICE, A Dataset of Drug-like Molecules and Peptides for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9813265/]
- 85. Chemical-Reactivity Properties, Drug Likeness, and ... [https://www.mdpi.com/2079-3197/7/3/52]
- 86. A methodology for meta-model based optimization in ... [https://www.sciencedirect.com/science/article/abs/pii/S0378778811005962]
- 87. Blockchain-Integrated Secure Authentication Framework ... [https://www.mdpi.com/1424-8220/25/21/6622]
- 88. Federated intelligence for smart grids: a comprehensive ... [https://jesit.springeropen.com/articles/10.1186/s43067-025-00235-8]
- 89. The convergence of pharmacometrics and quantitative ... [https://www.sciencedirect.com/science/article/pii/S0928098723000118]
- 90. Nanoparticles and convergence of artificial intelligence for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9853978/]
- 91. Master Protocols for Drug and Biological Product ... [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/master-protocols-drug-and-biological-product-development]
- 92. The Convergence Advantage: A Framework for Integrating ... [https://www.drugpatentwatch.com/blog/the-convergence-advantage-a-framework-for-integrating-patent-clinical-and-scientific-data-to-drive-life-sciences-innovation-strategy/?srsltid=AfmBOoouEvkVkzVjse6URNVX4Dcf7pdzc0qPgFO3pPflSrX6VdYKTXOm]